
記住我


The prevalence of diabetes mellitus (DM) is increasing worldwide, and type 2 diabetes mellitus (T2DM), characterized by hyperglycemia and relative insulin deficiency, is the major type of DM. T2DM can lead to multiple organ damage including eyes, kidneys, peripheral nerves, and peripheral blood vessels, if not treated appropriately and timely.
One of major complications linked to T2DM is low skeletal muscle mass, that is, muscular atrophy. Skeletal muscle comprises 45% of total body weight and 50% to 75% of body proteins and is closely related to energy metabolism.1 Skeletal muscle is the main regulators of glucose metabolism and a major site of insulin resistance in patients with T2DM.2 Muscular dystrophy leads to low skeletal muscle mass, which is independently associated with an increased risk of T2DM.3,4 In fact, low skeletal muscle mass and T2DM have a bidirectional relationship. For example, T2DM patients are associated with higher risks of sarcopenia,5,6 a chronic complication of T2DM,7 and low skeletal muscle mass can also influence the quality of life of T2DM patients, and is closely related to physical function decline, impaired cardiopulmonary function, infection, metabolic disorders, mobility disorders, fractures, and disabilities.8,9 In addition, low muscle mass is independently associated with all-cause mortality of T2DM patients.10
Liraglutide, a glucagon-like peptide-1 receptor (GLP-1R) agonist, improves glycemic control by stimulating the secretion of insulin and inhibiting the secretion of glucagon,11 and reduces body weight by inhibiting appetite center and food intake and delaying gastric emptying of food.12 Additionally, liraglutide offers many other beneficial effects, such as reducing blood pressure,13 improving lipid,14 decreasing the risk of cardiovascular disease,15 and slowing albuminuria progression.16 Previous studies suggested that GLP-1R agonists may benefit skeletal muscle mass and function.17 For example, liraglutide improved mitochondrial function and myofibril injury in skeletal muscle of T2DM patients,18 and the latest research showed that GLP-1R agonists ameliorated skeletal muscle atrophy.19,20 However, the mechanisms behind the beneficial effects of liraglutide on skeletal muscle in T2DM patients are not fully understood.
It is well known that skeletal muscle loss, regardless of the causes, involves decreased protein synthesis and increased protein decomposition in muscle, which is in part caused by activation of the ubiquitin protease system. The ubiquitin protease system is a three-enzyme cascade mediated by the sequence of E1 ubiquitin activating enzyme, E2 ubiquitin conjugating enzyme, and E3 ubiquitin ligase,21 where the target proteins are recognized by specific ubiquitin ligase E3 and then degraded. Atrogin-1 (MAFbx) and MuRF1 are specific ubiquitin proteases E3 for muscle atrophy,22 and their levels are elevated during hyperglycemia.23,24 Thus, the breakdown of skeletal muscle proteins is increased in diabetic patients. Therefore, MAFbx and MuRF1 are important regulators of skeletal muscle mass. However, whether there is any functional link between GLP-1R activation and the activity of MAFbx and MuRF1 has not been well investigated.
In the present study, we performed both in vivo and vitro experiments to investigate the molecular basis by which liraglutide ameliorates muscular atrophy. We found that liraglutide achieved its beneficial effects on skeletal muscle mass at least in part through inhibiting MAFbx and MuRF1 activity.
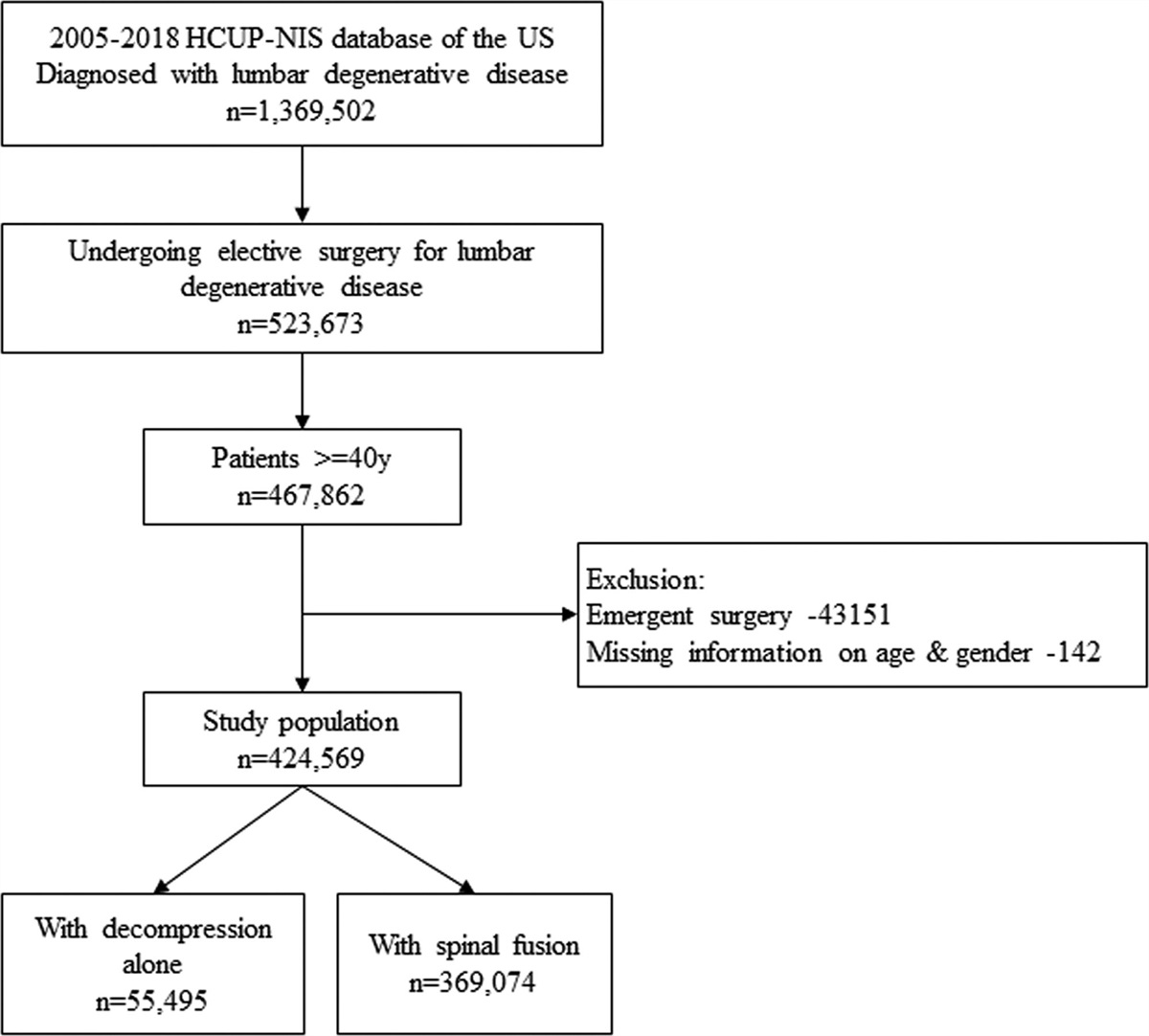
2. METHODS 2.1. Human subjectsThis study recruited 51 nonobese male T2DM patients (age range: 45–70 years), who had been admitted to the First Hospital of Qinhuangdao, Hebei, China, for glycemic control treatment between March 2020 and February 2021. Dual-energy X-ray absorptiometry (DXA) was used to assess skeletal muscle index (SMI) of all subjects. Among these 51 patients, 25 with a SMI <7 kg/m2 were assigned in the experiment group, and 26 with a SMI ≥7 kg/m2 were in the control group. SMI was identified as the ratio of skeletal lean mass to the square of body height in meters. All subjects in two groups were matched by age, body mass index (BMI), and serum levels of glycosylated hemoglobin (HbA1c), and were treated with noninsulin, non-GLP-1, and non-DPP4 methods. The criteria for T2DM diagnosis were in accordance with World Health Organization (WHO) 1999: having a fasting plasma glucose (FPG) level ≥7.0 mol/L and 2-hour postprandial blood glucose level ≥11.1 mol/L. Patients who had one of the following were excluded from this study: sports-related occupations, peripheral neuropathy, disused muscle atrophy, anemia, malignant tumor, hepatopathy, end-stage renal disease, thyroid dysfunction, autoimmune disease, severe cognitive disorder, arthrophlogosis, carpal tunnel syndrome, cervical disc herniation with compression of nerves, long-term bedridden, failed to complete dual-energy X-ray absorptiometry or had a history of stroke, heart stent, artificial heart pacemaker, or other metal implants implanted in the body. Patients who had addicted to alcohol in the past 2 years, taken special nutritional supplements such as protein powder, vitamin D, and calcium in recent 3 months, or changed their lifestyle and diet structure in recent 3 months were also excluded. In addition, patients who received treatments of insulin, GLP-1R agonists, DPP4 and drugs that affected blood sugar (such as glucocorticoids) were excluded from the study. This study was approved by the Ethics Committee of the First Hospital of Qinhuangdao. Written informed consent was obtained from all subjects prior to enrollment.
2.2. Anthropometric measurementsEach subject’s height and weight were measured using an electronic scale (HGM-800; Henan Shengyuan Industrial Co., Ltd.). During all measurements, subjects wore light clothing and were barefoot. Blood pressure levels, that is, systolic blood pressure (SBP) and diastolic blood pressure (DBP), were measured twice with an electronic sphygmomanometer (HBP-9020; Omron, Osaka, Japan) after 10 minutes of rest while the subjects were seated. The average of 2 measurements was used for analysis. BMI was calculated as weight (kg) divided by height squared (m2). Total body muscle mass, skeletal muscle mass, and fat mass of participants were measured by dual-energy X-ray absorptiometry (MEDIX90 France). SMI = Skeletal muscle mass (SMM) (kg)/height (H)2(m2). SMI is a diagnostic indicator of reduced skeletal muscle mass. According to AWGS, the cut off of SMI for reduction of muscle mass is less than 7.0 kg/m2 (male).25
2.3. Measurements of biochemical indices and GLP-1 and DPP4Levels of fasting blood glucose (FBG), and plasma lipids, including total cholesterol (TC), total triglycerides (TG), low-density lipoprotein cholesterol (LDL-c), and high-density lipoprotein cholesterol (HDL-c), were measured using the glucose oxidase-phenol 4-aminoantipyrine peroxidase method and enzymatic colorimetric assays with a biochemical auto-analyzer (Hitachi 7600 automated analyzer, Tokyo, Japan). Glycosylated hemoglobin (HbA1c) levels were measured using high performance liquid chromatography with an auto-analyzer (HA-8180, ARKRAY Factory, Inc., Japan). Serum GLP-1 and DPP4 levels were measured by enzyme-linked immunosorbent assay for quantitative detection kits (NBP2-82126, NOVUS, USA; DC260B, R&D SYSTEMS, USA).
2.4. AnimalsTen diabetic male mice, that is, KK-Ay mice (11- to 12-week old), were purchased from the animal house of Beijing Huafukang Technology Co., Ltd. Mice were maintained in the animal facility at a temperature of 22 ± 2°C and a humidity of 50 ± 5% on a 12-hour light and 12-hour dark cycle in pathogen free (SPF) environment, and given a high-fat diet (10% lard, 20% sucrose, 1% gallbladder salt, 2.5% egg yolk, 66.5% conventional feed) and water ad libitum. These mice were divided into two groups based on treatments: the liraglutide (LG) group (n = 5), in which mice were subcutaneously injected with liraglutide (Novo Nordisk, Denmark) 250 µg/kg/d for 8 weeks, and the saline group (control, n = 5), in which mice were subcutaneously injected with an equal volume of physiological saline. Food intake, blood glucose, and blood ketones were monitored every 3 days and body weight every week for a total of 8 weeks. The animal protocol was approved by the Laboratory Animal Science Research Committee of Nankai University, Tianjin.
2.5. Skeletal muscle histologyAfter 8 weeks of treatment, all mice fasted overnight, blood was obtained from eyeball after anesthesia by ether followed by cervical dislocation. Skeletal muscle tissues of the limbs were collected, and the wet weight was obtained. The muscles of hind legs were immediately frozen in liquid nitrogen and then frozen at −80°C for subsequent analysis such as polymerase chain reaction (PCR) and Western blot (WB).
The fat mass of hind limb of the mice was measured by Dual-Energy X-ray Absorptiometry (XR-600, NORLAND, Midmark Corporation, USA).
2.6. Blood glucoseFBG levels were measured by glucose oxidase method (Shanghai Rongsheng, China).
2.7. Real-time PCRTotal RNA was extracted from the frozen skeletal muscle using Trizol/chloroform procedure (Vazyme, Nanjing, China), and the RNA concentration and quality was determined by NanoDrop Meter. Total RNA (1 µg) was reverse transcribed into complementary DNA (cDNA) by using SuperMix (Yeasen, Shanghai, China) in an ABI-Prism 7200 (Applied Biosystems). The quantitative PCR was performed with the Light Cycler96 real-time PCR system (Roche, Switzerland). The ΔCT value was obtained by subtracting the CT value of the internal control β-actin from the CT value of the target gene. All expression of target genes of interest was normalized to β-actin. The sequences of primers for detecting genes of interest are listed in Table 1.
Table 1 - Sequences of primers used in this study Gene Forward (5′-3′) Reverse (5′-3′) β-actin CATGTACGTTGCTATCCAGGC CTCCTTAATGTCACGCACGAT MuRF1 (Trim63) GTGTGAGGTGCCTACTTGCTC GCTCAGTCTTCTGTCCTTGGA Atrogin-1 (Fbxo32) CAGCTTCGTGAGCGACCTC GGCAGTCGAGAAGTCCAGTCProteins were extracted from skeletal muscles using RIPA Lysis Buffer (Cwbio, Beijing, China) with an ultrasonic disintegrator, separated by 10% SDS-poly-acrylamide gel electrophoresis (PAGE), and transferred to polyvinylidene difluoride (PVDF) membrane. The membrane was incubated with the primary antibody of interest at 4°C overnight, followed by incubation with the corresponding horseradish peroxidase (HRP)-conjugated secondary antibody at 4°C for 2 hours. Specific protein bands were visualized using Chemiluminescence kit (Millipore, Billerica, MA, USA) and detected with Light-Capture Tanon-5200 (Yuanpinghao Biotechnology Co., Ltd. Beijing, China). The primary antibodies used in the Western blot were β-actin, MAFbx, and MuRF1 (Abcam, Cambridge, MA, USA).
2.9. Myogenic differentiation in vitroC2C12 cells (Shanghai Cell Bank of Chinese Academy of Sciences, Shanghai, China) were maintained in DMEM containing 20% fetal bovine serum (FBS) (growth medium, GM) supplemented with 1% penicillin and streptomycin. To induce myogenic differentiation, C2C12 cells were placed in coverslips and reached 95% confluence in GM, and then GM was replaced by DMEM containing 2% horse serum (differentiation medium, DM) supplemented with 1% penicillin and streptomycin for 5 days. To evaluate the effects of liraglutide on myogenic differentiation, at differentiation day 1, vehicle or liraglutide (different concentrations: 10, 100, and 1000 nM, respectively) was added into the medium until differentiation day 5. Myosin heavy chain (MHC) staining was used to evaluate the differentiation efficiency.
2.10. In vitro myotube atrophyIn vitro myotube atrophy was induced by pretreatment of C2C12 cells with dexamethasone (DEX) as previously reported but with slight modification.26 Briefly, C2C12 cells were pretreated with 10 µM DEX for 6 hours, followed by differentiation induction for 2 days. After then, C2C12 cells were divided into the following three groups: vehicle, Dex+vehicle, Dex+ligraglutide (10 nM), Dex+ligraglutide (1000 nM). C2C12 cells were further differentiated for 3 days.
2.11. Generation of adenovirusesAdenoviruses (Ad) expressing GFP as a control, MAFbx, and MuRF1 were generated (GeneChem, Shanghai, China) and infected C2C12 cells for 48 hours at a MOI of 100. Viral transduction efficiency was evaluated by GFP. After 48 hours infection, C2C12 cells were induced differentiation and treated with Dex and ligraglutide as mentioned earlier.
2.12. Immunofluorescence stainingC2C12 cells were seeded to coverslips, and fixed in 4% paraformaldehyde (Beyotime, Shanghai, China) for 10 minutes at room temperature. Cells were incubated with MF20 antibody (14-6503-82, Thermo Fisher Scientific, Shanghai, China) at 4°C overnight, followed by incubation with an Alexa Fluor 594 antibody (ab150076, Abcam, Shanghai, China) for 1 hour at room temperature. The 4´, 6-diamidino-2-phenylindole (DAPI, Beyotime, Shanghai, China) was used as nuclear staining. Immunofluorescence was captured by a fluorescent microscope.
2.13. Statistical analysisAll analyses were performed using the SPSS statistical software (SPSS for Windows, Version 24.0 SPSS Inc., Chicago, IL, USA). Normally distributed variables were expressed as means ± standard deviation (SD). Non-normally distributed variables were expressed as medians (interquartile range, IQR). Differences between groups were evaluated by using Pearson’s chi-square test or Student’s t test as applicable. The associations of SMI with various parameters were determined using univariate analysis and multiple linear regression analyses models, with SMI as the dependent variable, and age, duration, BMI, FBG, HbA1c, GLP-1, and DPP4 as independent variables. p < 0.05 was considered statistically significant.
3. RESULTS 3.1. T2DM patients with a lower SMI had significantly lower GLP-1 levels than T2DM patients with a higher SMIA total of 51 male T2DM subjects (n = 51) were included in our study and divided into two groups based on SMI: the experiment group with a SMI < 7 kg/m2 (n = 25), and the control group with a SMI ≥7 kg/m2 (n = 26). There were no significant differences between these two groups with regard to demographic and basal clinical characteristics including age, gender, FBG, DPP4, TG, TC, LDL-c, and HDL-c (Table 2). However, the experiment group had significantly lower GLP-1 levels than the control groups (p < 0.05). Univariate analysis revealed that SMI was positively correlated with GLP-1 (r = 0.526, p < 0.05) (Table 3), and multiple linear regression analyses revealed that SMI were positively associated with GLP-1 (β = 0.435, p = 0.001) and inversely associated with age (β = 0.299, p = 0.015) after adjustment for BMI, FBG, DPP4, TG, TC, LDL-C, and HDL-C (Table 4). Multiple logistic analyses showed that if GLP-1 levels were grouped by quartiles, the incidence of low muscle mass below the second quartiles was 10.55 times that of above the second quartiles (odds ratio = 10.556, p < 0.001). Thus, GLP-1 levels are positively correlated to SMI in T2DM patients.
Table 2 - Comparison of demographic and basal clinical characteristics of subjects between experimental and control groups Variables Experiment n = 25 Control n = 26 t p Age (y) 60.8 ± 9.03 59.0 ± 7.85 0.775 0.442 Duration (y) 10.6 ± 8.64 7.4 ± 6.43 1.494 0.142 BMI (Kg/m2) 23.7 ± 2.87 24.8 ± 1.40 −1.671 0.104 FBG (mmol/L) 7.9 ± 2.51 8.0 ± 2.69 −0.175 0.862 HbA1c (%) 7.8 ± 1.43 8.6 ± 2.59 −1.443 0.157 SMI (kg/m2) 6.3 ± 0.5 7.5 ± 0.38a −9.795 <0.001 GLP-1 (ng/mL) 0.3 ± 0.22 0.5 ± 0.20a −3.770 <0.001 DDP4 (ng/mL) 411.0 ± 211.22 429.2 ± 141.70 −0.36 0.721 TG (mmol/L) 2.28 ± 2.8 1.79 ± 0.8 0.825 0.417 TC (mmol/L) 5.10 ± 1.3 5.26 ± 1.0 −0.478 0.635 LDL-c (mmol/L) 2.88 ± 0.9 3.09 ± 0.8 −0.904 0.37 HDL-c (mmol/L) 1.28 ± 0.3 1.25 ± 0.2 0.379 0.706Comparisons were performed between the two groups using Chi-square test or student’s t test as appropriate.
ap < 0.05 was considered statistically significant.
BMI = body mass index; DPP4 = Dipeptidyl-Peptidase IV Inhibitors; FBG = fasting blood glucose; GLP-1 = glucagon-like peptide-1; HbA1c = Glycosylated hemoglobin; HDL-c = high-density lipoprotein cholesterol; LDL-c = Low-density lipoprotein cholesterol; SMI = Skeletal muscle index; TC = total cholesterol; TG = triglyceride.
ap< 0.05 was considered statistically significant.
BMI = body mass index; DPP4 = Dipeptidyl-Peptidase IV Inhibitors; FBG = fasting blood glucose; GLP-1 = glucagon-like peptide-1; HbA1c = Glycosylated hemoglobin.
ap< 0.05 was considered significant.
GLP-1 = glucagon-like peptide-1.
We next used the mouse DM model to study the effects of liraglutide, a GLP-1R agonist, on muscular atrophy. DM was induced by high-fat diet in KK-Ay mice. These animals were then divided into two groups, the liraglutide (LG) group, in which mice were treated with liraglutide 250 µg/kg/d for 8 weeks, and the control group, in which mice were treated with the same volume of physiological saline (NaCl) for 8 weeks. Liraglutide-treated mice had significantly lower food intake, body weight, and fasting blood glucose levels, but higher wet weight of limbs’ skeletal muscle mass (Table 5 and Fig. 1). Also, liraglutide-treated mice had significantly lower expression of MuRF1 and MAFbx in skeletal muscles compared with NaCl-treated mice as revealed by RT-qPCR (p < 0.05) (Fig. 2A). Consistent with the qPCR results, the protein levels of MuRF1 and MAFbx protein expression in LG group were significantly lower than in control group (Fig. 2B).
Table 5 - Effects of liraglutide on food intake, body weight and laboratory characteristics of DM mice Valuable Wt (g) Food Intake (g) FBG (mmol/L) SMM (g) Fat Mass (Hind Limb) (g) KK-Ay-NaCl (n = 5) 40.7 ± 2.74 112.1 ± 14.27 33.3 ± 5.82 1.6 ± 0.11 1.6 ± 0.34 KK-Ay-liraglutide (n = 5) 37.1 ± 1.93 94.2 ± 12.99 20.9 ± 3.81 1.9 ± 0.16 1.4 ± 0.29 p <0.01a 0.001a 0.03a 0.001a 0.276ap < 0.05 was considered significant.
Wt = body weight; FBG = fasting blood glucose; SMM = skeletal muscle mass.
 Fig. 1:
Fig. 1: Liraglutide treatment improves body weight and food intake of diabetic mice. **p < 0.05, ***p < 0.01, LG vs. Cont at the respective time point (week).
 Fig. 2:
Fig. 2: Liraglutide treatment decreases the expression of MuRF1 and MAFbx in muscles of diabetic mice as revealed by RT-qPCR (A) and Western blot (B). B, actin serves as an internal control. n = 5 per group. *p < 0.05.
3.3. Liraglutide promoted myogenic differentiationTo further examine the effects of liraglutide on skeletal muscle biology, we used C2C12 cell line, a widely used in vitro system for skeletal muscle study, to probe the effects of liraglutide on myogenic differentiation. First, C2C12 cells were maintained in GM, and incubated with vehicle, or different concentrations of liraglutide (10, 100, and 1000 nm, respectively), for 5 days. As shown in Fig. 3A, liraglutide promoted myogenic differentiation in a dose-dependent manner. Similarly, liraglutide reduced the expression of MAFbx and MuRF1 transcription in myogenic cells in a dose-dependent manner (Fig. 3B). Consistent with the above observations, liraglutide promoted myogenic differentiation of C2C12 cells cultured in DM for 5 days, and reduced the expression of MAFbx and MuRF1 as well (Fig. 4A,B). Taken together, we conclude that liraglutide promotes myogenic differentiation at least in vitro.
 Fig. 3:
Fig. 3: Liraglutide promotes myogenic differentiation and decreases the mRNA expression of MuRF1 and MAFbx in myogenic cells. A. C2C12 cells were maintained in GM, and treated with vehicle, and different concentrations of liraglutide (10, 100, 1000 nM) for 5 days. MHC staining was performed to evaluate the differentiation. DAPI is a nuclear staining. Magnification, 20×. Bar, 100 µm. B. RT-qPCR was performed to evaluate the effects of liraglutide on the mRNA expression of MuRF1 and MAFbx. *p < 0.05, **p < 0.01, ***p < 0.001, vs. vehicle group.
 Fig. 4:
Fig. 4: Liraglutide promotes myogenic differentiation and decreases the mRNA expression of MuRF1 and MAFbx in differentiating cells. A. C2C12 cells were maintained in GM to reach 95% confluency, then induced to differentiate in DM for 5 days. Vehicle or different concentrations of liraglutide (10, 100, 1000 nM) started treatment at the day of medium switch and lasted for 5 days. MHC staining was performed to evaluate the differentiation. DAPI is a nuclear staining. Magnification, 20x. Bar, 100 µm. B. RT-qPCR was performed to evaluate the effects of liraglutide on the mRNA expression of MuRF1 and MAFbx. **p < 0.01, ***p < 0.001, vs. vehicle group.
3.4. Liraglutide ameliorated DEX-induced myotube atrophy in vitroMyotube atrophy was induced by incubation of 2-day differentiating C2C12 cells with DEX. These cells were then treated with vehicle, liraglutide 10 and 1000 nM, respectively. As shown in Fig. 5A, while vehicle only group showed myotube formation, indicating differentiation, DEX treatment significantly reduced it. Liraglutide ameliorated DEX-induced myotube atrophy in a dose-dependent manner. DEX treatment also substantially increased the levels of MAFbx and MuRF1, which was reduced by liraglutide treatment (Fig. 5B). Collectively, we argue that liraglutide ameliorates DEX-induced myotube atrophy in vitro, which is accompanied by reduction in the expression of MAFbx and MuRF1.
 Fig. 5:
Fig. 5: Liraglutide attenuates DEX-induced myotube atrophy and decreases the DEX-induced mRNA expression of MuRF1 and MAFbx. A. C2C12 cells were induced differentiation for 2 days, and then treated with DEX and vehicle, liraglutide 10 and 1000 nM for 3 days. Vehicle alone serves as a control. MHC staining was performed to evaluate the differentiation. DAPI is a nuclear staining. Magnification, 20x. Bar, 100 µm. B. RT-qPCR was performed to evaluate the effects of liraglutide on the mRNA expression of MuRF1 and MAFbx. *p < 0.05, **p < 0.01 or ***p < 0.001 vs. vehicle group. ##p<0.01 vs. Dex+vehicle group. &&p<0.01 vs. Dex+ liraglutide (low dose) group.
3.5. Ectopic expression of MAFbx and MuRF1 antagonized the beneficial effects of liraglutide on DEX-induced myotube atrophyWe next examined whether decreased expression of MAFbx and MuRF1 contributed to the beneficial effects of liraglutide on DEX-induced myotube atrophy. We generated Ad-GFP as a control, Ad-MAFbx and Ad-MuRF1, and transduced them in C2C12 cells for 48 hours at a MOI of 100. Fig. 6A shows the transduction efficiency with these Ads. After then, differentiation was induced for two days, followed by treatment with either vehicle or DEX to induce myotube atrophy. Liraglutide (1000 nM) were then added into these cells for 3 days. Myotube atrophy was evaluated by MHC staining. DEX treatment significantly impaired myotube formation, which was ameliorated by liraglutide (Fig. 6B). However, Ad-MAFbx and Ad-MuRF1 significantly antagonized the amelioration of myotube formation by liraglutide (Fig. 6C), indicating that liraglutide attenuates DEX-induced myotube atrophy at least partially through suppressing the expression of MAFbx and MuRF1.
 Fig. 6:
Fig. 6: Ectopic expression of MAFbx and MuRF1 antagonizes the beneficial effects of ligraglutide on Dex-induced myotube atrophy. A. Evaluation of adenoviral transduction efficiency as revealed by GFP. B. Experiments were performed as described in Figure 5A, except that GFP, MuRF1, and MAFbx were transduced by adenoviruses for 48 hours before Dex pretreatment. MHC staining was performed to evaluate the differentiation. DAPI is a nuclear staining. Magnification, 20x. Bar, 100 µm. C. RT-qPCR was performed to evaluate the effects of liraglutide on the mRNA expression of MuRF1 and MAFbx. *p < 0.05, **p < 0.01, ***p < 0.001, vs. vehicle+Ad-GFP.
4. DISCUSSIONIn the present study, we first examined the serum GLP-1 levels in two groups of nonobese T2DM patients based on a SMI < 7 kg/m2 and a SMI ≥7 kg/m2, and found that the T2DM patients with a lower SMI had significantly lower GLP-1 levels than those with a higher SMI. We next tested the effects of liraglutide, a GLP-1R agonist, on the mouse DM model, and found that liraglutide treatment significantly decreased body weight and improved skeletal muscle mass of DM mice, which was accompanied by decreased expression of MuRF1 and MAFbx in skeletal muscles. Our in vitro myogenic differentiation studies further revealed that liraglutide promoted myogenic differentiation, attenuated myotube atrophy, and suppressed atrophy-induced increase in mRNA expression of MuRF1 and MAFbx. However, ectopic expression of MuRF1 and MAFbx antagonized the beneficial effects of liraglutide on myotube atrophy, suggesting that liraglutide improves skeletal muscle mass in atrophy at least in part through suppressing the expression of MuRF1 and MAFbx.
Our study suggested that GLP-1 was a protective factor of SMI while age was a risk factor of SMI after adjustment for BMI, FBG, DPP4, TG, TC, LDL-C, and HDL-C. When GLP-1 levels were grouped by quartiles, the incidence of low muscle mass below the second quartiles was 10.55 times that of above the second quartiles. Low muscle mass in T2DM patients was reported to be linked to a variety of factors, including age, hyperglycemia, IGF-1 level, insulin resistance, muscle fat infiltration, inflammatory factors, peripheral neuropathy, and mitochondrial damage,27-29 and that low muscle mass increased the risk of diabetes.3,4 Actually, muscle mass gradually and slowly decreases starting from the middle age, but accelerates obviously after age of 75.30 In T2DM patients, both leg muscle mass and muscle strength may decrease by more than 30% in 3 years compared with nondiabetic patients.31 On the other hand, a Korean study also showed that hyperglycemia is negatively related to muscle strengths and quality.32 While people pay more attention to muscle strength, it is also proposed that sarcopenia, the reduction of muscle strength, muscle mass, and function,25 is a chronic complication of diabetes. In the present study, we observed that the endogenous GLP-1 level of nonobese middle-aged T2DM patients decreased in coincidence with decrease in the muscle mass, the latter of which was independent of blood glucose. Thus, whether GLP-1 contributed to the development of T2DM-linked muscular atrophy is not clear.
We used diabetic mice (KK-Ay), a popular T2DM animal model,33 to interrogate the involvement of GLP-1 in muscular atrophy. The age of mice at 19–20 weeks was considered to be the middle age, thus excluding the influence of old age. The mice treated with liraglutide had reduced food intake, body weight, and FBG, while the wet weight of limbs’ skeletal muscle mass was significantly increased, which coincided with the decreased expression of MuRF1 and MAFbx. Thus, activating GLP-1 by liraglutide, a GLP-1R agonist, exerted beneficial effects on the muscle mass of diabetic mice, which was consistent with the previous findings that GLP-1 has a protective effect on the loss of muscle mass in diabetes in both patients and animal models.34,35
Previous studies from the same research group have shown that GLP-1R activation by Ex-4, a GLP-1R agonist, and dulaglutide, a long-acting GLP-1R agonist, improved muscle atrophy in coincidence with decreased levels of MAFbx and MuRF1.19,36 Our study used liraglutide, another long-acting GLP-1R agonist, and independently corroborated previous observations. Our study also suggested that the similar mechanisms probably held true in the treatment of diabetic muscle atrophy by liraglutide. On the other hand, liraglutide was shown to ameliorate skeletal muscle atrophy through protein kinase A and B signaling pathways as well as cAMP-dependent signaling cascades.19,20 It will be of great interest to completely decipher the signaling pathways that underpin the therapeutic effects of liraglutide on diabetic atrophy.
The mechanisms by which liraglutide improves skeletal muscle mass have been previously studied, and several potential mechanisms have been proposed. Liraglutide was shown to alleviate mitochondrial injure and muscle fiber damage in the muscles of diabetic patients by inhibiting oxidative stress, thereby maintaining muscle mass.18 On the other hand, liraglutide reduces inflammatory cytokines levels, decreases fat accumulation and eases insulin resistance, thus maintaining muscle mass in diabetic patients.37 Indeed, correcting hyperglycemia can effectively relieve muscle loss.38 Hence, hypoglycemic effect imposed by liraglutide is definitely one of the mechanisms to improve muscle loss in diabetic muscle atrophy. However, in our study, we did not observe a significant correlation between GLP-1 and HbA1c levels. While our small sample size potentially accounted for this lack of correlation, we stratified the HbA1c levels for association analysis but also did not find any significant correlation between them. On the other hand, insulin resistance is a basic component of the pathophysiology of muscle mass reduction,39,40 and diabetic patients have insulin resistance, increased levels of oxidative stress and release of proinflammatory factors. Liraglutide may reduce hyperglycemia and visceral fat, which in turn relieves insulin resistance. However, the T2DM patients enrolled in this study were nonobese, thus eliminating the influence of the reduced fat tissues on liraglutide-linked muscle benefits. To support the above conclusion, we did not observe any significant difference in fat mass of hindlimbs between vehicle- and liraglutide-treated diabetic mice.
Previously, liraglutide was found to promote myogenesis in cultured C2C12 cells.20 Indeed, we also found that liraglutide promoted myogenic differentiation in a dose-dependent manner. Moreover, in the m
留言 (0)