Patient samples
A total of 240 NSCLC and paired para-carcinoma tissues were available from patients who underwent surgical resections at the First Affiliated Hospital of Huzhou University between January, 2015 and December, 2018. Among them, 189 pairs of formalin-fixed paraffin-embedded (FFPE) tissues were consisted of a NSCLC tissue microarray (TMA) and analyzed using immunohistochemistry (IHC) staining, and 51 pairs of fresh tissues were used for reverse transcription-quantitative PCR and western blotting analyses. These patients were not received chemotherapy or radiation therapy superior to surgical resection. This research was approved by the Ethics Committee of the First Affiliated Hospital of Huzhou University (approved number: 2021KYLL007). Informed consents were obtained from all patients. All patients clinicopathological characteristic was shown in Additional files Table S1.
TCGA datasets
Four hundred seventy-two samples from 1 GEO datasets (GSE32863 [18]), Hou Lung and Okayama Lung [19, 20] were used to analyze the STRIP2 expression in NSCLC compared with normal tissues. In addition, GSE32863 dataset was used to analyze the IGF2BP3, P300 and CBP expressions in NSCLC. Two published IGF2BP3 RIP-sequence data [21] and GEO database (GSE90684) [21] were analyzed.
Kaplan-Meier plotter
The Kaplan-Meier plotter (www.kmplot.com) [22] was used to predict the relationship between STRIP2 mRNA expression and overall survival as well as recurrence-free survival. STRIP2 expression levels in patients with lung adenocarcinoma (n = 601) were collected using Kaplan-Meier Plotter online bioinformatics datasets. Additionally, patients were divided into low or high STRIP2 expression groups, according to STRIP2 mRNA expression based on the median value. The hazard ratio (HR) with 95% confidence intervals (CI) and the log-rank P value were calculated in the study.
Cell culture
Human normal bronchial epithelial cell line (BEAS-2B) and NSCLC cell lines (SPCA1, H1299, A549, PC-9, H1975, H23 and H226) were purchased from the Shanghai Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, cat. no. L110KJ, BasalMedia, Shanghai) or RPMI-1640 (cat.no. L210KJ, BasalMedia, Shanghai) supplemented with 10% fetal bovine serum (FBS, Gibco, Thermo Fisher Scientific), 100 U/ml penicillin and 100 μg/ml streptomycin (Sigma-Aldrich, Merck KGaA) in a humidified incubator under 5% CO2 at 37 °C.
Plasmid construction and cell transfection
Overexpression plasmids were cloned and constructed into a pcDNA3.1/myc-his (cat.no. V800–20, Invitrogen, Thermo Fisher Scientific, Inc.) or pcDNA3.1-3xFlag (VT8001, Youbio company, Hunan) or pXJ40-Flag (Our laboratory constructed) or pGEX-4 T-1 vector (VT1253, Youbio company, Hunan). For gene knockdown, sh-control (referred to as shCtrl), STRIP2 shRNAs (shSTRIP2#1/2) and IGF2BP3 shRNAs (shIGF2BP3#1/2) were designed and synthesized by Tsingke biological Technology (Hangzhou, China). In addition, two specific small interfering (si) RNAs of P300/CBP/TMBIM6 (20 μM) and scrambled siRNA (referred to as si-Ctrl; 20 μM) were designed and synthesized by Guangzhou RiboBio Co., Ltd. Cells were transfected with Lipofectamine® 2000 reagents (Invitrogen, Thermo Fisher Scientific, Inc.) according to the manufacturer’s instructions. Stable cell lines were selected using puromycin (cat.no. A1113803, Gibco, Thermo Fisher Scientific, Inc.). The primer sequences used in the experiment were as follows: shSTRIP2#1 targeted sequence, 5′-GGAACAAGTTCATCGGATT − 3′; shSTRIP2#2 targeted sequence, 5′-GCCGGAGCTTACTACTGAA-3′; shIGF2 BP3#1 targeted sequence, 5′-GCTGAGAAGTCGATTACTA-3′; shIGF2BP3#2 targeted sequence, 5′-TCGGAAACTTCAGATACGA-3′. si-P300#1 forward, 5′-CG UGUCUUCACUUAAGAGUdTdT-3′ and reverse, 5′-ACUCUUAAGUGAAGACA CGdTdT-3′; si-P300#2 forward, 5′-CAUACUUAGACGUUUGUUAdTdT-3′ and reverse, 5′-UAACAAACGUCUAAGUAUGdTdT-3′; si-CBP#1 forward, 5′-GUCAC CCUUGGAACAAGGUdTdT-3′ and reverse, 5′-UGGAACAAGGUUCCCACUGdT dT-3′; si-CBP#2 forward, 5′-CCUGACGUCAGAUGUACU dTdT-3′ and reverse, 5′-UCAUGAUAGACUGCAGUCCdTdT-3′; si-TMBIM6#1 forward, 5′-GUGGAAG GCCUUCUUUCUAdTdT-3′ and reverse, 5′-UAGAAAGAAGGCCUUCCACdTdT − 3′; si-TMBIM6#2 forward, 5′-UUCCGUGACGUAACUAGAGdTdT-3′ and reverse, 5′-CUCUAGUUACGUCACACGGAAdTdT-3′; and si-Ctrl forward, 5′-UUCUCCG AACGUGUCACGUTT-3′ and reverse, 5′-ACGUGACACGUUCGGAGAATT-3′.
Cell proliferation assay
Cell proliferation was detected using CCK-8 assay (Beyotime Institute of Biotechnology, Shanghai, China) and Real-time Cellular Analysis (RTCA assay; ACEA Biosciences, Inc.; Agilent Technologies, Inc.), as previously described [23,24,25]. For CCK-8 assay, cells were transfected with the plasmids and seeded in 96-well plate at a density of 5, 000–10, 000 cells/well. Cells were cultured for 24, 48, and 72 hours, followed by incubation with 10 μl CCK-8 reagent, the absorbance values at 450 nm were determined by a Spectra Max 190 reader (Molecular Devices, LLC). For RTCA assay, 5, 000–10, 000 cells were seeded into the E-plate. The data were recorded using xCELLLigence software 2.0 (ACEA Biosciences, Inc.; Agilent Technologies, Inc.) and analyzed using GraphPad Prism 5.0 (GraphPad Software, Inc.).
Transwell assay
The migratory and invasive abilities of NSCLC cells were analyzed using Transwell assay (Coring; pore, 8 μm; USA). In brief, cells were seeded in the upper chamber with serum-free medium, while the lower chamber contained complete medium to induce migration and invasion. Following the indicated time incubation, the migrated cells were fixed with 4% paraformaldehyde (PFA) and then stained with 1% crystal violet stain solution (Beyotime Institute of Biotechnology). For cell invasion, the upper chamber was firstly coated with Matrigel® Matrix Basement Membrane (cat. no. 356234; BD Biosciences, USA). The images were captured under an inverted fluorescence microscope (ZEISS Axio Vert.A1; Carl Zeiss AG). In addition, the stained cells were decolorized with 30% acetic acid, and the absorbance values were determined by a Spectra Max 190 reader (Molecular Devices, LLC) at 570 nm.
Real-time cellular analysis (RTCA)
The RTCA xCELLLigence system (ACEA Biosciences, Inc.; Agilent Technologies, Inc.) was usually used to measure cell morphology, proliferation and migration in vitro in a non-invasive manner, as described previously [23,24,25]. In brief, the lower chamber was added into complete medium, while cells were seeded into serum-free culture medium to induce migration and invasion. The data were recorded using xCELLLigence software 2.0 (ACEA Biosciences, Inc.; Agilent Technologies, Inc.) and analyzed using GraphPad Prism 5.0 (GraphPad Software, Inc.).
RNA extraction and RT-qPCR
Total RNA was extracted from tissues or cells using TRIzol® reagent (Invitrogen, Thermo Fisher Scientific, Inc.), according to the manufacturer’s instructions. 500 ng total RNA was reverse transcribed into cDNA using the PrimeScript RT reagent kit (Takara Biotechnology Co., Ltd.), as our previously described [24, 25]. RT-qPCR was performed using UltraSYBR Green PCR Master mix (cat. no. CW0957H; CWBIO, Beijing, China) on an ABI 7500 system (Applied Biosystems; Thermo Fisher Scientific, Inc.). The results were normalized to 18S ribosomal RNA (18sRNA), and the relative expression level was calculated using the 2-ΔΔCq method [26]. Sequences of the primers are described in Additional file: Table S2.
Western blot analysis
Total protein was extracted from tissues or cells using RIPA buffer (cat. no. P0013B; Beyotime Institute of Biotechnology) containing protease and phosphatase inhibitors (Beyotime Institute of Biotechnology). An equal amount of protein was loaded and separated with 8–12% SDS-PAGE. The protein was transferred onto a PVDF membrane (Invitrogen, Thermo Fisher Scientific, Inc.) and then incubated with primary antibodies at 4 °C overnight. The membrane was washed with phosphate buffer saline (PBS) containing 0.1% Tween-20 and incubated with corresponding second antibodies at room temperature for 1 hour. The images were obtained using the Tanon 5200 (Tanon).
GST fusion protein pull-down assay
The GST-vector and GST-IGF2BP3 fusion protein plasmids were transfected into Escherichia coli BL-21 (DE3) cells. Fusion proteins were induced expression by isopropyl β-D-thiogalactoside (IPTG) and purified using the GST protein purification kit (Beyotime Institute of Biotechnology) according to the manufacturer’s protocol. The bound protein was identified using Coomassie staining. GST fusion protein solutions were incubated with beads at 4 °C for 3 h. Whole 293 T cell lysates transfected with STRIP2-flag protein were incubated with beads containing GST fusion proteins at 4 °C overnight. The beads were washed and the protein was detected using western blotting assay.
Immunofluorescence
Cells seeded on coverslips were fixed in 4% PFA for 30 min at room temperature followed by permeabilization with 0.1% of Triton-X-100 (Beyotime Institute of Biotechnology) in PBS for 20 min at room temperature. After three time washing, fixed cells were then blocked with 5% Bovine Serum Albumin (BSA) in PBS for 1 hour at room temperature and incubated with a polyclonal rabbit anti-STRIP2 antibody (1:200, cat. no. PA5–54047, Invitrogen, Thermo Fisher Scientific, Inc.) at 4 °C overnight in darkness. Cells were washed with PBS and then incubated with goat-anti rabbit secondary antibody conjugated to Alexa Fluor 488 (Thermo Fisher Scientific, Inc.) for 1 hour at room temperature in darkness. Cells were stained using antifade solution containing DAPI and mounted on an inverted fluorescence microscope (ZEISS Axio Vert.A1; Carl Zeiss AG).
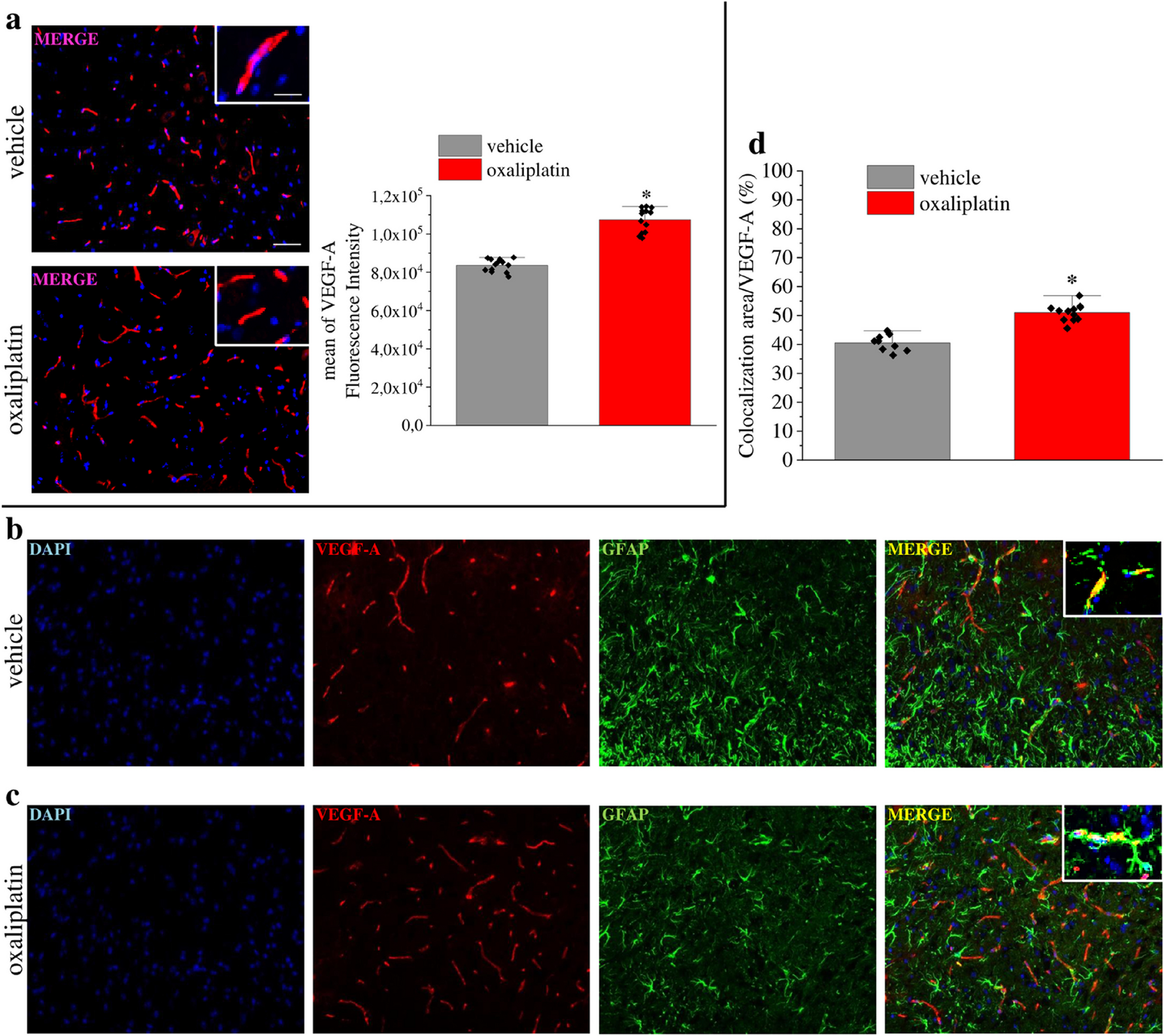
Immunohistochemistry staining
IHC staining was performed as described previously [25]. In brief, slides were incubated with anti-STRIP2 antibody (dilution 1:200), anti-IGF2BP3 antibody (dilution 1:200) and anti-TMBIM6 antibody (dilution 1:200) at 4 °C overnight and then cultured with SP Rabbit & Mouse HRP-conjugated secondary antibody (cat no. CW2096S; CWBio; https://www.cwbio.com/) for 1 h at room temperature. And the slides were incubated with DAB kit (cat. no. CW2096S; CWBio) following counterstained with hematoxylin staining solution (cat. no. C0107; Beyotime Institute of Biotechnology). The images were digitally scanned at a magnification of × 400 using a KF-PRO-005 platform (Ningbo Jiangfeng Bio-information Technology Co., Ltd.) into whole slide digital images. The scoring of the slides was conducted using HALO Multiplex IHC analysis software version v3.1.1076.308 (Indica Labs). The intensity was scored as 1 (absent or weak), 2 (moderate) or 3 (high). The percentage of positive cells was assigned as 1 (< 25%), 2 (25–50%) and 3 (> 50%). The score of each slice was multiplied to acquire a final score of 1–9.
Methylated RNA immunoprecipitation-PCR (MeRIP-qPCR)
The MeRIP-qPCR assay was conducted using the riboMeRIP™ m6a Transcriptome Profiling kit (Guangzhou RiboBio Co., Ltd.; Guangzhou), according to the manufacturer’s instructions. In brief, total RNA was extracted with TRIzol® reagent (Invitrogen, Thermo Fisher Scientific, Inc.) and RNA was fragmented using the RNA fragmentation buffer supplied with the kit. The specific anti-m6A antibody was vortically pre-bound to Protein A/G magnetic beads at room temperature for 30 min. Then the fragmented RNA was incubated with m6A-antibody-bound Protein A/G magnetic beads at 4 °C for 2 h and washed twice using wash buffer. The m6A-antibody-bound RNA was purified using GeneJET RNA Purification kit (Invitrogen, Thermo Fisher Scientific, Inc.). Real-time PCR was performed following m6A-IP to quantify the changes to m6A methylation of TMBIM6. The primer sequences used in the experiment were as follow: TMBIM6 5′-TCATATAACCCCGTCAACGC-3′ (sence) and 5′-CAAATCCAGCAAGAAGTC CC-3′ (antisence).
Chromatin Immunoprecipitation (CHIP) assay
CHIP assay was carried out using the SimpleChIP® Enzymatic Chromatin IP kit (Magnetic Beads) (cat. no. 9003; Cell Signaling Technology) according to the manufacturer’s protocol. Briefly, about 5 × 107 NSCLC cells were cross-linked with 1% formaldehyde for 10 min at room temperature. The cross-linking was quenched by addition of 125 mM glycine for 5 min at room temperature and followed by sonication. The fragmented chromatin was analyzed on agarose gels. After purifying, the chromatin was incubated with specific CBP antibody (cat. no. 7425; Cell Signaling Technology) or P300 antibody (cat. no. 54062; Cell Signaling Technology) to immunoprecipitation chromatin overnight at 4 °C with rotation and followed by incubation with protein G magnetic beads for 2 h at 4 °C. The eluted DNA was purified according to the kit and then used as template for RT-qPCR with specific primers: STRIP2 (promoter), 5′-CCCAGTCATGGGCTGCA ATAA-3′ (sense) and 5′-CTGAGCACAGAAGTCACCAGTT-3′ (antisense). The control primers for human RPL30 were provided with the kit. The process of RT-qPCR was carried out according to described previously [27, 28].
RNA and protein stability assays
To explore the stability of TMBIM6 mRNA and protein under the influence of downregulation or upregulation of STRIP2/IGF2BP3, cells were treated with actinomycin D (5 μg/ml; cat. no. HY-17559; MedChemExpress) for 0, 6, 12, 24 h. The procedures of total RNA isolation and RT-qPCR were performed as described previously [29].
RNA sequencing
Total RNA was harvested and isolated from the stable STRIP2 knockdown and scramble control cells. The total RNA was then performed to ribosomal RNA depleted RNA sequencing (RNA-Seq) protocols as previously described [30, 31]. The library was sequenced on an Illumina Novaseq 6000. Differentially expressed genes were defined as fold change > 2 or fold change < 0.5 and p < 0.05, and then Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed. In addition, the venn diagram was conducted using STRIP2 knockdown sequence data and two published IGF2BP3 RIP-sequence data and GEO database (GSE90684). All protocols and analyses were provided by LC Biotech Corporation (Hangzhou, China).
Luciferase reporter assay
Luciferase reporter assay was performed as previous study [29]. In brief, cells were transfected with a luciferase reporter containing TMBIM6 WT or Mut and Renilla plasmids. Luciferase activities were detected using a Dual-Luciferase Reporter Gene Assay Kit (Beyotime Institute of Biotechnology).
Cell apoptosis assay
Cell apoptosis assay was carried out using the Annexin-V/propidium iodide (PI) detection kit (cat. no. PF00005; Proteintech Group, Inc.,), according to the manufacturer’s protocol. In briefly, cells were transfected with scrambled siRNA and two specfic siRNAs of TMBIM6 for 24 hours. After transfection, the cells were harvested and washed with phosphate buffer saline (PBS) twice and then resuspended in a binding buffer containing Annexin-V and PI for 15 min on ice in darkness. Cells were measured using the BD FACSCanto II system (BD Biosciences).
Statistical analysis
All data were represented as mean ± SEM of three independent experiments and analyzed using SPSS software version 21.0 (IBM Corp.) with an unpaired Student’s t-test or one-way ANOVA followed by a Tukey’s post hoc test. The associations between STRIP2 expression and patient clinicopathological features of patients were analyzed using the χ2 test and Fisher’s exact test. The correlations of STRIP2, IGF2BP3 and TMBIM6 expression were analyzed using Pearson rank correlation analysis. Kaplan-Meier survival analysis was performed using the log-rank test. P value< 0.05 was considered statistically significant.






留言 (0)