
記住我
Among the 337 patients, 7 patients from the SPLC group were pathologically diagnosed with squamous cell carcinoma (n = 5) and large-cell neuroendocrine carcinoma (n = 2), and 7 patients were found to have lymph node metastasis at the time of surgery (3 patients in SPLC group and 4 patients in sMPLC group). These 14 patients were excluded from the following analyses. In all, it included 191 patients with 191 lung nodules in the SPLC group and 132 patients with 295 lung nodules in the sMPLC group. All lesions were no larger than 3.0 cm in the greatest dimension verified by pathological reports. Nodules from sMPLC patients showed significant genetic heterogeneity. Most patients with multifocal adenocarcinomas were histopathologically different or harbored different genetic alternations. Nevertheless, about 8.3% (11/132) of the patients in the sMPLC group had the same gene mutation profiles. CT features and histopathological analyses were re-evaluated for these patients to confirm the sMPLC diagnosis [5, 9]. Based on CT findings, pathological results, and follow-up data, all 323 patients were diagnosed as T1-2N0M0 (stage Ia-Ib) SPLC or sMPLC patients. (Some tumors were T2 based on visceral pleural invasion and not size.) Clinical and demographic variables are shown in Additional file 1: Table S1.
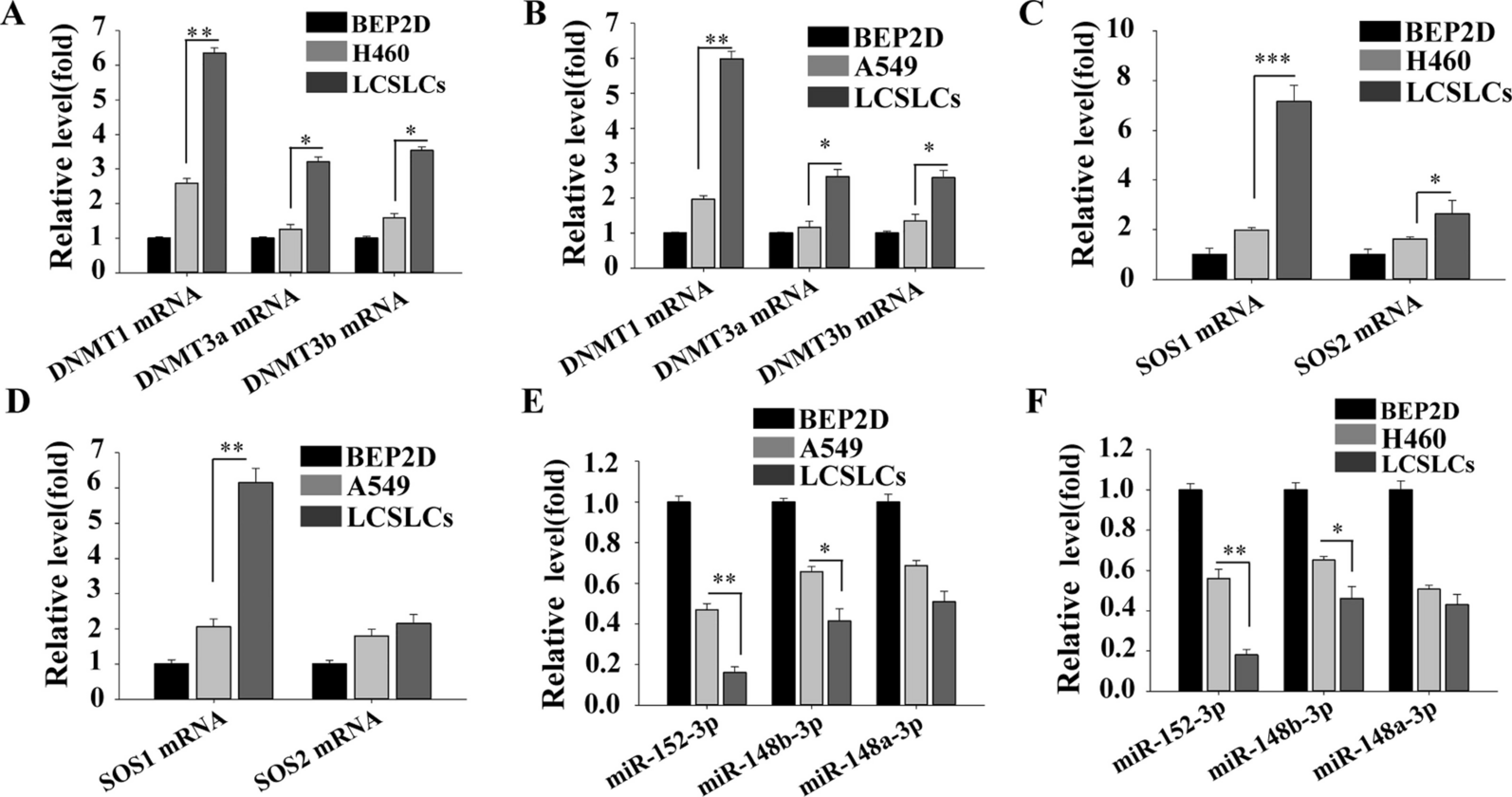
The tumor mutation burden in SPLC and sMPLC patientsWe first investigated the tumor mutation burden (TMB) status. The changes in TMB showed similar trends in both groups. TMB was higher in older patients and gradually increased parallel to the tumor size (Additional file 2: Table S2). In terms of CT features, TMB gradually increased from ground-glass opacity (GGO) lesions to mixed GGO (mGGO) and solid lesions (Fig. 1B and C), while in terms of pathological subtypes, TMB was significantly higher in invasive adenocarcinoma (IAC) lesions than that in adenocarcinoma in situ (AIS)/minimally invasive adenocarcinoma (MIA) lesions in both groups (Additional file 2: Table S2).
Fig. 1
Tumor mutation burden in SPLC and sMPLC patients. A Generally, sMPLC lesions had lower TMB levels than SPLC nodules. B and C in both SPLC and sMPLC patients, TMB gradually increased from GGO lesions to mGGO and solid lesions. D and E When compared between the two groups, the TMB of GGO and mGGO lesions from sMPLC patients was significantly lower than that of SPLC patients. F The solid nodules from sMPLC patients also showed a similar trend, but no statistical significance was found. *, p < 0.05; **, p < 0.01; ***, p < 0.001
We then investigated the TMB differences between the two groups. In terms of patient age, TMB of sMPLC lesions showed a lower trend than SPLC lesions in all age-groups, despite statistical difference only being found in the subgroup of 40- to 60-year-old patients (p < 0.0001). Although the TMB was similar between the two groups in the nodules less than 1 cm, patients from the SPLC group had significantly higher TMB levels when the tumor size was larger than 1 cm (Additional file 2: Table S2). Interestingly, when compared between the two groups, the TMB of GGO and mGGO lesions from sMPLC patients was significantly lower than that of SPLC patients (Fig. 1D and E, Additional file 2: Table S2). The solid nodules from sMPLC patients also showed a similar trend, but no statistical significance was found (Fig. 1F). The TMB had no differences in AIS/MIA lesions between the two groups, but in IAC nodules, sMPLC patients had significantly lower TMB levels than SPLC patients. Our study demonstrated that sMPLC lesions had lower TMB levels than those from SPLC nodules (Fig. 1A, Additional file 2: Table S2).
Gene mutation landscape in SPLC and sMPLC patientsGene mutational analyses were performed on all the lesions included in this study. In both groups, a higher frequency of EGFR mutation was found in females (69.4% in the SPLC group and 63.0% in the sMPLC group), while KRAS, LRP1B, FAT1, GRIN2A, KEAP1, STK11, TP53, PIK3CG, RET, and RB1 were more mutated in males (Additional file 3: Table S3A and F). The MED12 and FGFR3 mutations were more detected in younger patients, and ARID1A was more mutated in older cases (Additional file 3: Table S3B). In the sMPLC group, the ERBB2, NOTCH4, KCNQ2, and XRCC3 mutations were more found in younger patients and mutated RBM10, TP53, LRP1B, PTPRD, and STK11 were more detected in the patients more than 60 years old (Additional file 3: Table S3G). Similar to previous studies in both groups, the gene mutation rate significantly increased as the tumor became larger, except for ERBB2, which was more mutated in smaller lesions (Additional file 3: Table S3C and H). When comparing the gene mutation among GGO, mGGO, and solid lesions, the EGFR mutation was detected in the SLPC group (61.7% of GGO lesions, 70.0% of mGGO lesions, and 41.3% of solid nodules) and in the sMPLC group (44.8% of GGO lesions, 65.8% of mGGO lesions, and 79.2% of solid nodules). The mutations of RET, PIK3CG, and PDGFRA were only found in solid nodules. In the SPLC group, the mutation frequency of TP53, KRAS, PTPRD, KEAP1, SMAD4, STK11, RB1, and GRIN2A became higher as the consolidation part of the lesion increased (Additional file 3: Table S3D). The mutation rate of TP53, MAP3K1, IFNG, MYC, CTNNB1, PIK3CG, MDM2, and NOTCH2 significantly increased from GGO lesions to solid lesions in sMPLC patients (Additional file 3: Table S3I). The TP53 gene mutation was the most enriched alteration in the IAC compared to AIS/MIA lesions. The MED12, BRCA1, and SOX17 mutations were found more in AIS/MIA lesions (Additional file 3: Table S3E). When investigating the significantly mutated genes according to histological features in the sMPLC group, the mutation rate of TP53 was significantly higher in IAC lesions, and the MAP2K1, BCOR, MED12, MST1R, and BRAF mutations were more enriched in MIA/AIS nodules (Additional file 3: Table S3J).
Genetical discrepancies between SPLC and sMPLC patientsThe gene mutational discrepancies were investigated between SPLC and sMPLC patients. In GGO lesions, the EGFR mutation rate was lower in the sMPLC group than that in the SPLC group (44.8% vs. 61.7%, p < 0.05, Fig. 2A), while BRAF mutation was more frequently observed in sMPLC cases (Fig. 2A). Gene mutation status had no differences between the two groups in mGGO lesions (Fig. 2B). Interestingly, the EGFR mutation was significantly higher in solid lesions from the sMPLC group (79.2% vs. 41.3%, p < 0.01, Fig. 2C), which was the opposite of the GGO lesions.
Fig. 2
Genetical discrepancies between SPLC and sMPLC patients-CT feature oriented. A In GGO lesions, the EGFR mutation rate was lower in the sMPLC group than that in the SPLC group (44.8% vs. 61.7%, p < 0.05), while BRAF mutation was more frequently observed in sMPLC cases (18.1% vs. 5.0%, p < 0.05). B Gene mutation status had no differences between the two groups in mGGO lesions. C In solid lesions, the EGFR mutation was significantly higher in the sMPLC group than in SPLC patients (79.2% vs. 41.3%, p < 0.01)
We further analyzed the gene mutation differences in subgroups regarding gender, age, nodule size, and CT features. The gene mutation rate did not differ between the two groups in nodule size. In female patients with GGO lesions, BRAF mutation was more found in the sMPLC group (16.3% vs. 2.4%, p = 0.033), while APC (0.0% vs. 7.1%, p = 0.036) and EGFR (47.5% vs. 67.0%, p = 0.037) mutation rate was higher in SPLC group (Additional file 4: Table S4). Most patients in this study were between 40 and 60 years old. In this age period, the patients with multiple GGO lesions harbored more BRAF (18.6% vs. 2.5%, p = 0.022) mutation and less APC (0.0% vs. 10.0%, p < 0.01) mutation (Additional file 5: Table S5B). A higher frequency of TP53 mutation of mGGO lesions was found in SPLC patients (30.0% vs. 13.0%, p = 0.028, Additional file 5: Table S5E). In the patients with solid nodules, EGFR mutation was significantly enriched in the sMPLC group (81.0% vs. 38.0%, p = 0.0067, Additional file 5: Table S5H).
Overall changes in DNA methylation patterns in sMPLC and SPLCGenome-wide analyses of DNA methylation were conducted, and the density plot showed relatively similar methylation levels in the samples from the sMPLC and SPLC (Additional file 9: Fig. S1A). Principal component analysis (PCA) based on all CpG sites did not reveal any discernable separation between the two groups (Additional file 9: Fig. S1B). The heatmap and cluster analysis showed that, in sMPLC, unlike the high heterogeneity in the genome, the DNA methylation patterns were very similar among the multiple lesions from the same patient. Based on the genome-wide DNA methylation patterns, the sMPLC and SPLC tumor samples could be directly clustered into two groups, indicating that the DNA methylation patterns were different between the two groups (Fig. 3A and B).
Fig. 3
Genome-wide DNA methylation patterns in sMPLC and SPLC. A The heatmap and cluster analysis showed that, in sMPLC, unlike the high heterogeneity in the genome, the DNA methylation patterns were very similar among the multiple lesions from the same patient. B Based on the genome-wide DNA methylation patterns, the sMPLC and SPLC tumor samples could be directly clustered into two groups. C The DNA methylation level of SPLC tumors around the transcription starting site (TSS) was significantly higher than that in the sMPLC tumors. D and E DMP identified in sMPLC and SPLC tumors, compared to their normal lung tissues. The genes corresponding to the top 20 differences in DMP are listed at the two tips of the volcanic map. F DMP identified between sMPLC and SPLC tumors. The genes corresponding to the top 20 differences in DMP are listed at the two tips of the volcanic map. MC, multiple primary lung cancers; SC, single primary lung cancer
A total of 3364 differentially methylated positions (DMP) were found in sMPLC tumors (1936 hypomethylated and 1428 hypermethylated), and 20,444 DMP were found in SPLCs (11,995 hypomethylated and 8449 hypermethylated), compared to their normal lung tissues, respectively. These substantially methylated sites appear to be bimodal distribution and randomly distributed on 22 chromosomes (Additional file 9: Fig. S1C and D). The distribution percentages of the DMP on different gene regions were slightly different between sMPLC and SPLC (Additional file 9: Fig. S1E and F). The genes corresponding to the top 20 differences in DMP are listed at the two tips of the volcano plot (Fig. 3D and E). GO and KEGG analyses revealed the DMP in sMPLC and SPLC involved in different biological processes, cellular functions, and pathways (Additional files 6 and 9: Table S6 and Fig. S1G, H).
The PPI network analysis identified 12 essential differentially methylated genes, and related epigenetic function modules were established as statistically significant in the sMPLC group. Among these genes, SPINT2, LNX1, FOXA2, and SOSTDC1 were reported to be related to tumor progression, whereas TYROBP, TREM2, IL23R, CSF2RB, TRAF1, and TGFBR1 were closely associated with immune response. The genes with up‐regulated or down‐regulated methylation levels had complex interactions in the signaling network (Additional file 7: Table S7).
In the SPLC, 15 essential differentially methylated genes and epigenetic function modules were identified. PPI network analysis revealed these genes and modules were mainly related to tumor progression (LOXL1, IKBKE, LNX1, FOXA2, FGF17, and FGF2) and immune response (STAT4, IL23R, LTA, ITK, and IL19). LNX1, FOXA2, and IL23R overlapped between sMPLC and SPLC, while other modules indicated that the DNA methylation patterns in sMPLC were different from SPLC. The enrichment results are presented in detail in Additional file 7: Table S7.
DNA methylation discrepancies between sMPLC and SPLCWe next investigated the DNA methylation discrepancies between the two groups by directly comparing the DNA methylation status between the tumor samples from sMPLC and SPLC. Interestingly, the DNA methylation pattern around the transcription starting site (TSS) differed between the two groups. And the methylation level of SPLC tumors around TSS was significantly higher than that in the sMPLC (Fig. 3C, Additional file 9: Fig. S2B and C). A total of 8068 DMP (3201 hypomethylated and 4867 hypermethylated) were identified. Heatmap generated from clustering analysis illustrating DMP between sMPLC and SPLC, and the genes corresponding to the top 20 differences in DMP are listed at the two tips of the volcano plot (Figs. 3F and 4A). GO functional analysis showed that these DMP are involved in different biological processes, cellular components, and molecular functions (Additional file 9: Fig. S2A). The top 20 prominently enriched KEGG pathways of the DMP are presented in Table 1. The most significant pathway was the cholinergic synapse pathway (p = 3.08E-06). The pathway with the greatest number of DMPs was the chemokine signaling pathway (23 DMPs, p = 1.33E-05).
Fig. 4
DNA methylation discrepancies between sMPLC and SPLC. A Heatmap generated from clustering analysis illustrated the DMP between sMPLC and SPLC. B and C The PPI network analysis identified CXCR2, EDN1, and ADRA1A as essential differentially methylated genes. The epigenetic function modules were established to illustrate the interactions of the related genes. The CXCR2 and ADRA1A closely interacted and were shown in a same module. D and E Aberrant DNA methylation at the TSS region was significantly related to the abnormal expression of immune-related genes. In sMPLC, the r = − 0.541, p = 0.002; in SPLC, the r = − 0.773, p < 0.0001
Table 1 Top 20 prominently enriched KEGG pathways of the DMP between sMPLC and SPLCThe PPI network analysis identified three essential differentially methylated genes, CXCR2, EDN1, and ADRA1A, and related epigenetic function modules were established to illustrate the interactions of the related genes (Fig. 4B and C). The CXCR2 and ADRA1A closely interacted, and the signaling network of related genes indicated that the function module might involve in the immune response regulation in the tumor microenvironment (Table 2).
Table 2 Essential differentially methylated genes and the genes in the epigenetic function modulesImmune characterization of SPLC and sMPLC patientsIn the SPLC group, 30 immune-related DEGs were identified with 15 genes down-regulated and 15 genes up-regulated (Fig. 5A and B, Additional file 8: Table S8). The significant GO terms and KEGG pathways analyses showed that these DEGs were related to multiple biological processes, molecular functions, and pathways involved in immune response regulation (Additional file 9: Fig. S3A and C). Interestingly, our data identified more immune-related DEGs in sMPLC patients (50 genes down-regulated and 8 genes up-regulated). Only 14 DEGs were shared between the two groups (Fig. 5B and C, Additional file 8: Table S8). Different from SPLC patients, more immune-related biological processes and pathways, such as B cell activation, B cell receptor signaling pathway, and IL-17 signaling pathway, were identified in the sMPLC group (Additional file 9: Fig. S3B and D). Visualizing biomolecular interaction networks [20] are shown in Fig. 5D and E. Moreover, we investigated the correlation between DNA methylation and gene expression. Aberrant DNA methylation at the TSS region was found in most of these abnormal expressed immune-related genes, indicating that DNA methylation may involve in the immune response regulation of early-stage lung cancer (Fig. 4B and C).
Fig. 5
A, B and C In SPLC patients, 30 immune-related DEGs were identified with 15 genes down-regulated and 15 genes up-regulated. In sMPLC patients, 58 immune-related DEGs were identified with 50 genes down-regulated and 8 genes up-regulated. Fourteen DEGs were shared in both groups. D and E Visualizing biomolecular interaction network in SPLC and sMPLC. F CIBERSORT analyses revealed immune cell infiltrating differences between SPLC and sMPLC patients. In both groups, tumor tissues showed more significant infiltration of naïve B cells, TfH cells, Tregs, activated NK cells, resting dendritic cells, and macrophages. Moreover, in the sMPLC group, memory B cells, plasma cells, and activated mast cells were significantly more infiltrated in tumor tissues than normal lung tissues. Interestingly, a certain group of immune cells (B cells, dendritic cells, Tregs, and activated memory CD4+ T cells) showed significant infiltrating differences even in normal lung tissues between SPLC and sMPLC patients. *, p < 0.05; **, p < 0.01; ***, p < 0.001. sMPLC, tumors from sMPLC group; sMPLC-N, normal lung tissues from sMPLC group; SPLC, tumors from SPLC group; SPLC-N, normal lung tissues from SPLC group
We then evaluated the TILs in tumor and non-tumor tissues from SPLC and sMPLC patients. In both groups, tumor tissues showed more significant infiltration of naïve B cells, TfH cells, Tregs, activated NK cells, resting dendritic cells, and macrophages. Moreover, in the sMPLC group, memory B cells, plasma cells, and activated mast cells were significantly more infiltrated in tumor tissues than in normal lung tissues (Fig. 5F). Immunofluorescence analyses showed similar results to the CIBERSORT (Additional file 9: Fig. S3E). Together, our data demonstrated the existence of immune profile discrepancies between SPLC and sMPLC patients. DNA methylation may involve immune dysregulation in the tumor microenvironment of SPLC and sMPLC.
留言 (0)