
記住我
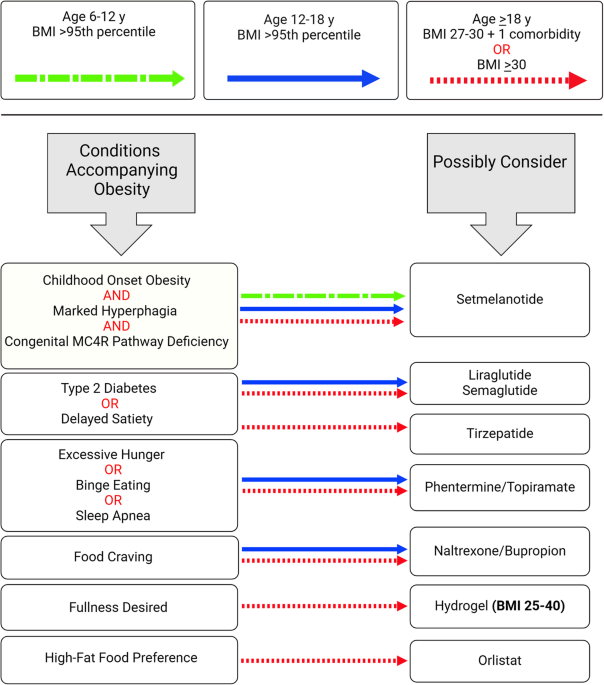


Clinical characteristics of the participants are detailed in Supplementary Table S1. Body mass index, blood pressure, and lipid serum levels did not differ significantly between the OD and OND groups. As expected, the OD group had significantly (p value <0.05) higher HbA1c (6.3 ± 1% vs. 5.4 ± 0.2%) and serum glucose levels (7.49 ± 4.3 mmol/l vs 4.53 ± 0.52 mmol/l) compared with the OND group.
Differential methylation between OD and OND groupsTo identify DNA methylation differences between the OD and OND groups, we compared VAT methylation profiles obtained with Illumina EPIC microarrays. We found DMCs in all chromosomes. The highest and longest density of epigenomic alterations was observed on chromosome 6, with 132 DMCs encompassing 70 genes at the Major Histocompatibility Complex (MHC). Other regions showing high densities of alterations were located on chromosomes 4 and 11 (Supplementary Fig. S1). In the OD group, we found 11 120 DMCs (5880 genes), of which 48.4% were hypomethylated and 51.6% were hypermethylated compared to the OND group (Fig. 1, Supplementary Table S2). An unsupervised hierarchical cluster analysis and multi-dimensional scaling of the DMCs showed a clear methylation profile for each patient group (Fig. 1b, Supplementary Fig. S2).
Fig. 1: Comparison of visceral adipose tissue DNA methylation profiles between patients with obesity but without diabetes (OND) and patients with obesity and diabetes (OD).
a Volcano plot shows differences in methylation. Points represent all analyzed CpGs, and blue points indicate DMCs (FDR < 0.05): lighter blue hypomethylated and darker blue hypermethylated in OD, and gray points non-significant. b Heat map of DMCs showing DNA methylation levels for each CpG (row) by patient (columns), after applying an unsupervised hierarchical clustering analysis. c Distribution of the DMCs among the different genomic locations. d Distributions of DMCs across the CGI, the shore (2 kb from the CGI), the shelf (2–4 kb from the CGI), and the open sea (the remaining genome) regions. e Gene set enrichment analysis of DMCs (Affinity propagation). Kyoto Encyclopedia of Genes and Genomes pathway enrichments associated with genes with DMCs. X axis: FDR (false discovery rate) value. DMCs differentially methylated CpGs, TSS1500 1500–200 bp upstream of the transcriptional start site (TSS), TSS200 200 bp upstream of the TSS, IGR intergenic regions, UTR untranslated region, CGI CpG island.
Notably, DMCs that showed the highest delta β values (>20%) were associated with genes that were newly related to T2D (TRANK1, TEX2, SH2D3C, ATAD1, ANKEF1, MIR138-2, OR10A5, SIM1, PRRC2C, TECRL, ZDHHC14, PHTF1, C11orf66, SH3TC2, MRGPRX1, RNF212, and FLJ16171) or previously related to T2D (FSD1L, NSF, SLIT3, PTPRN2, PSMD10, MAD1L1, MIR572, ATM, LCLAT1, and TNFRSF8) (Supplementary Table S13). Among the 11 120 DMCs, 71% were intragenic, including 39.6% that were mainly distributed in the gene body and 31.4% that were in promoter regions, upstream of the TSS (Fig. 1c). Most DMCs were found in regions with low CpG content, like the shore (17.6%), shelf (7.1%), and open sea regions (58.5%), compared with CpG islands (16.8%; Fig. 1d).
Using the Bumphunter algorithm, we found 96 DMRs that were mainly hypermethylated (74%) in the OD compared to the OND group. Most of these DMRs had overlapping CpG-rich regions (CpG islands; Supplementary Table S3). Furthermore, 92 of the 96 DMRs were located within gene regions. Of these DMRs, 80 were in the vicinity of a TSS, including some that extended into the gene body, and 12 were confined to gene bodies. Additionally, overlaps between DMCs and DMRs were observed in 54 genes, the most significant being in BLCAP, SLC25A24, PM20D1, PAX8, and LCLAT1 (Fig. 2a and Supplementary Table S13).
Fig. 2: Overlap of differential methylation and gene expression.
a Venn diagram showing overlap between genes with DMCs or DMRs and the DEGs. b Gene set enrichment analysis of overlapping DMC-DEG genes. Kyoto Encyclopedia of Genes and Genomes pathway enrichments associated with genes with DMCs. X axis: p value. DMCs differentially methylated CpGs, DMRs differentially methylated regions, DEGs differentially expressed genes.
Among the genes with DMCs, the enrichment analysis mainly identified pathways related to fatty acid metabolism, aldosterone synthesis and secretion, the oxytocin signaling pathway, GABAergic synapse, and dopaminergic synapse, among others (Fig. 1e and Supplementary Table S4). After FDR correction, enrichment analysis of genes with DMRs was not able to identify any significantly enriched pathways.
Overlapping changes between DNA methylation and gene expression (DMC-DEG)Gene expression analysis identified 252 DEGs between the OD and OND, with 55.6% being overexpressed and 44.4% underexpressed in the OD (Supplementary Fig. S3 and Supplementary Table S5). Overlap between altered expression and methylation was observed with DMCs, but not with DMRs (Fig. 2a and Supplementary Table S6). Out of the 252 DEGs, 68 (DMCs = 88) showed altered methylation (DMC-DEG); in 35 it was located in the promoter region, and in 53 it was in the gene body. Among those genes with promoter DMCs, 12 were hypomethylated (8 overexpressed and 4 underexpressed) and 23 were hypermethylated (12 overexpressed and 11 underexpressed). In addition, among the genes with DMCs in the gene body, 25 were hypomethylated (8 overexpressed and 17 underexpressed) and 28 hypermethylated (16 overexpressed and 12 underexpressed). Enrichment analysis of the 68 overlapping genes did not reveal any significant pathways at FDR < 0.05. However, with a nominal p value (<0.05), we found important enriched pathways such as PPARG and Hippo signaling (Fig. 2b and Supplementary Table S7). Other important pathways were synthesis and degradation of ketone bodies; butonate metabolism; valine, leucine, and isoleucine degradation; and fatty acid metabolism, among others.
Correlation between methylation and gene expressionTo investigate correlation between expression and methylation, we matched the β-methylation values and expression levels of the DMC-DEG. Among the 88 DMCs (68 genes), we observed 26 (24 genes) with a significant DMC-DEG correlation (Supplementary Table S6). The top five correlations were observed in the genes ATP11A, LPL, PRRX1, ABCC9, and EHD2 (Fig. 3 and Supplementary Table S13).
Fig. 3: Correlations between differentially methylated CpGs, gene expression, and T2D-related traits.
a Plot displaying only the DMCs with significant Pearson correlation (p value <0.05) between DNA methylation levels and corresponding gene expression, fasting glucose, and HbA1c levels; blue squares indicate those with significant p values. b–d ATP11A: Pearson’s correlation for two of its representative DMCs with expression levels. Red dots represent OD patients, purple represents OND; the size of the dot depends on the value of each T2D-related trait. X axis: β-methylation; Y axis: ATP11A expression levels. b cg25043602-ATP11A expression: r = –0.533, p = 0.037; cg25043602-HbA1c: r = 0.686, p = 0.002. c cg25043602-Glucose: r = 0.539, p = 0.031; d cg16762784-ATP11A expression: r = −0.766, p = 0.003; cg16762784-HbA1c: r = 0.548, p = 0.038.
Identification of correlations between differential methylation and T2D-related traitsTo identify the potential relationships of DNA methylation with HbA1c and fasting glucose, we performed Pearson’s correlation analysis using CpG β-methylation values on genes showing DMC-DEG. From 88 DMCs, we found 38 (35 genes) significant correlations with HbA1c (p value <0.05), of which 11 were also correlated with fasting glucose (Supplementary Table S6). Notably, from the 24 genes with DMC-DEG correlation, the methylation of 16 genes, including ATP11A, LPL, EHD2, ACVR1C, and MAP4, was also significantly correlated with T2D-related traits (Fig. 3 and Supplementary Table S13).
Support of methylation analyses: validation and extended analysesTo support the methylation findings, we performed a validation analysis of the DMCs combining two female public datasets (11 OND and 10 OD) [13, 14]. When CpGs from the DMCs obtained in our initial study were contrasted between OND and OD included in the validation dataset, we observed 233 CpGs showing the same effect directions of differential methylation, at a nominal p value (<0.05) (Supplementary Table S8). These genes also enriched the glutamatergic synapse, long-term depression, and Hippo signaling pathways (Supplementary Table S9), similar to those observed in our sample.
Additionally, to gain statistical power in our findings, we performed a multi-ethnic extended analysis, combining the datasets contained in both public repositories with our own. When we compared the OD (n = 20) and the OND (n = 20) groups of the extended cohort, we found 9 648 DMCs in 5 135 genes (Supplementary Fig. S4 and Supplementary Table S10). Out of these, 2 092 genes and 945 DMCs were also found when our cohort was independently analyzed (Supplementary Table S2). All of the shared DMCs showed consistent directionality in both analyses, except for cg25140607 at TFAP2A (Supplementary Table S13). Similar to what was observed in our group of Mexican patients, in the multi-ethnic extended cohort the unsupervised hierarchical cluster analysis of the DMCs was able to separate OND and OD patients independently of their ethnic background (Supplementary Fig. S4b). Again, LCLAT1 displayed multiple DMCs and showed the highest delta β values (>25%), together with GSTTP2/GSTT1 (Supplementary Table S13). Furthermore, the enrichment analysis revealed 26 pathways shared between the two analyses, including oxytocin signaling, GABAergic synapse, glutamatergic synapse, Hippo signaling pathway, MAPK signaling pathway, circadian entrainment, aldosterone synthesis, and secretion, among others (Supplementary Table S11). In addition, 32 DMRs (GALR1, LCLAT1, SLC25A24, SLC1A2, GRIK2, TDRD12, MIR886, GSTO2, LRCOL1, etc.) were found in the same genes as the DMRs observed in our cohort (Supplementary Tables S12, S13).
留言 (0)