
記住我




As an aerobic muscular organ, the myocardium needs to be perfused with blood rich in oxygen, provided by the coronary circulation, for metabolism and contraction. Ischemic heart disease (IHD) occurs as a consequence of the imbalance between the demand and the supply of oxygenated blood to cardiac tissue; adult cardiac myocytes, in the condition of reduced oxygen, become incapable of maintaining intracellular metabolism, leading to cell apoptosis and necrosis and serious tissue damage. It should be noted that tissue injury strictly depends on ischemic duration, thus a major effort should be given to reduce the time of intervention and to assure a rapid onset of the reperfusion. Reperfusion, however, aggravates the damage, leading to activation of myocardial cell damage eventually resulting in uncontrolled cell death: this event is usually known as ischemia-reperfusion injury (IRI) and has substantial effects on the clinical outcome of the patient, especially because adult cardiac tissue is almost unable to regenerate after injury, resulting in a permanent damage (Araszkiewicz et al., 2013; Frank et al., 2012).
According to World Health Organization (WHO), cardiovascular diseases (CVDs) are the leading causes of death, IHD is the second global cause of disability-adjusted life years and the first global cause of death with an impact of 8,9 million of deaths in the world per year (https://www.who.int/data/gho/data/themes/mortality-and-global-health-estimates). Global Burden of Disease study estimated 197 million prevalent cases of IHD worldwide, causing 49,2% of total CVDs deaths (Roth et al., 2020). IHD is not only a disease of the old people in rich countries, but also several studies indicate an impact of this pathological condition on working-age adults and increasing trouble in low- and middle-income countries (Murray and Lopez, 1997; Abegunde et al., 2007). A positive fact is that the mortality due to IHD has declined over the past 4 decades in western countries thanks to significant progress in interventional procedures and medical treatments, to the reduction of risk factors and to lifestyle improvements.
As clinical definition, myocardial infarction (ST-segment elevation myocardial infarction (STEMI) and non-STEMI), stable angina and ischemic cardiomyopathy are part of the group of IHD (Heusch, 2019).
In fact, this type of disease is complex and multifactorial: several studies have linked IHD to environmental factors such as physical activity, stress, diet, smoking and to a variety of cardiovascular risk factors, as family history of cardiovascular disease, hypercholesterolemia, hypertension, or diabetes (Khera et al., 2016; Whayne and Saha, 2019). A not-modifiable risk factor is gender: there is a higher prevalence of IHD in men in comparison to women, in a way independent by age between the two genders but with an earlier age of onset in men (Khan et al., 2020).
Due to the impact of IHD on global health, its pathophysiology has been deeply studied and several efforts have been made to depict molecular pathways underlying its complexity in order to ameliorate clinical interventions and therapies. At present, there are not effective therapies in use focusing on reduction of IRI, thus the understanding of the molecular mechanisms underlying this unavoidable damage is fundamental in order to develop cardioprotective strategies. In particular, taking into account that mitochondria have an important role in IHD pathophysiology, recent efforts oriented toward the study of these organelles with the aim of ameliorating clinical approaches to treat IHD. It should be noted that, IRI is not a specific cardiac process, but it occurs in several other organs leading to a variety of diseases. For instance, IRI is an important complication after kidney and heart transplantation (Salvadori et al., 2015; Liu et al., 2018), leading to primary graft failure and reducing the survival of recipients. IRI has also been found as a common feature of ischemic stroke (L et al., 2016). Thus studies concerning IRI focused on the disentangling of the molecular mechanisms involved in its onset are still important in order to develop new therapies for this pathological process.
Cardiac tissue requires a large amount of energy to carry out its function, in particular for contractile capacity and for the control of electrical conduction (Murphy et al., 2016) and these processes are supported by the intensified mitochondrial network of cardiomyocytes. In fact, in cardiac myocytes mitochondria occupy almost 30% of the total volume. To note, in mammalian cells, mitochondria are primarily localized around the nucleus; on the contrary, in adult cardiomyocytes, mitochondria are highly organized and localized in three different areas among the cardiac fibers that can be evidenced with multimodal microscopy (Barzda et al., 2005). This highly coordinated localization is reached during the development, in fact, fetal mitochondria are not well organized, they lack well-formed cristae and of a specific localization (Garbern and Lee, 2021). The different localization of mitochondria in cardiomyocytes may be associated to different functions: subsarcolemmal (SSM), which has been suggested to supply ATP for the active transport of metabolites and electrolytes across the sarcolemma; perinuclear (PNM), with a not yet clear function but with a potential relation to nuclear metabolism; and intermyofibrillar mitochondria (IFM), which are the most active for oxidative metabolism and connected to muscle contraction (Shimada et al., 1984; Hollander et al., 2014).
Mitochondria are semi-autonomous organelles, highly motile and able to integrate and communicate with different compartments: in the last years, the direct contact between the mitochondria and other intracellular organelles has been deeply investigated, particularly with the endoplasmic reticulum (ER) (Angebault et al., 2020; Marchi et al., 2014). The mitochondria and ER contacts are among the most critical studied contact sites, probably because their occurrence is considered as a hub for Ca2+ homeostasis and the exchange of lipid membrane (Lee and Min, 2018). Nevertheless, communication between the mitochondria and other intracellular organelles has been observed in several studies, albeit the nature of all the contacts is still unclear (for details see Figures 1–3).
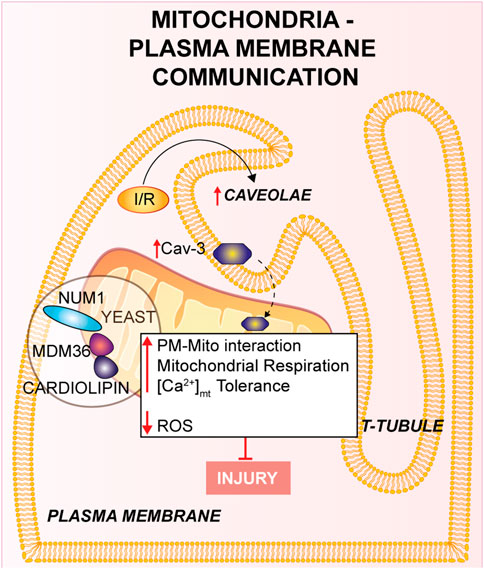
FIGURE 1. Mitochondria-Plasma membrane communication. Num1/Mdm36 complex anchors the PM to the OMM, with the involvement of cardiolipin. Cardiomyocyte’s T-tubules displays mitochondria-associated caveolae (PM invaginations), which number is increased in response to I/R. Furthermore, the overexpression of Cav-3 showed a cardioprotective effect against I/R.
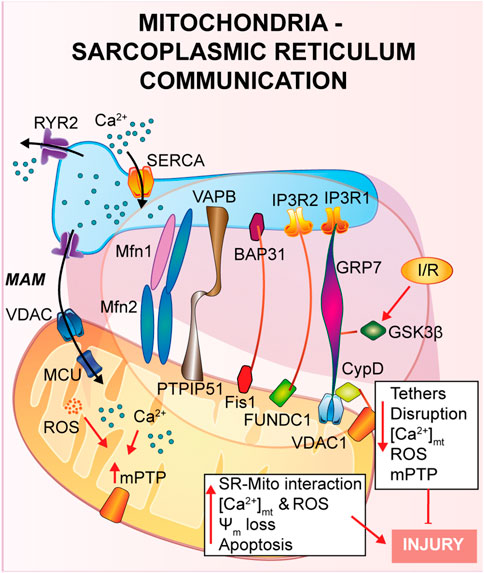
FIGURE 2. Mitochondria-Sarcoplasmic reticulum interaction. This type of interaction forms a region called MAM, fundamental for the presence of several proteins that guarantee a correct tethering and regulate Ca2+ cycle. In general, a reduction in the SR-mitochondria tethering correlates with decrease in mitochondrial Ca2+ uptake, reduction of cell death and cardioprotection to IRI.
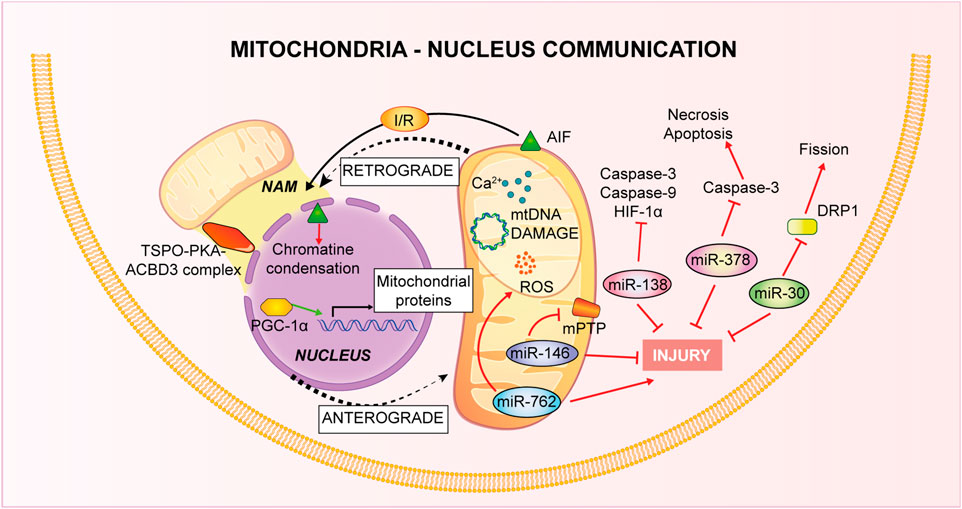
FIGURE 3. Mitochondria-Nucleus communication. Nucleus control of mitochondrial health is named anterograde signaling, through nuclear genes encoding mitochondrial proteins and via miRNAs. Mitochondria, conversely, use a retrograde signaling induced by Ca2+-dependent stress, mtDNA damage, metabolic disorders and loss of ψm.
Mitochondria’s plethora of roles includes ATP production through oxidative phosphorylation (OXPHOS), reactive oxygen species (ROS) production, programmed cell death control, intracellular Ca2+ homeostasis (Galluzzi et al., 2012). Thus, taken into consideration their importance, it is obvious that mitochondrial dysfunction is linked to the pathogenesis of different cardiovascular disease (Bonora et al., 2019; Pedriali et al., 2020). The mitochondrial involvement is of greatest interest especially for IHD. During ischemia the lack of oxygen stops the mitochondrial respiratory chain and the consequent ATP production (Hausenloy and Yellon, 2013), it leads to an intracellular Ca2+ overload that, at the moment of reperfusion, provokes Ca2+ buffering by mitochondria, increases oxidative stress, mitochondrial permeability pore (mPTP) opening, which in turn contributes to cardiomyocytes death (Morciano et al., 2015). Beyond inter-organelle interactions, mitochondria state of health is controlled by their morphology, with an extremely precise interchange of process of fusion and fission or biogenesis and mitophagy (as described in Figure 4).
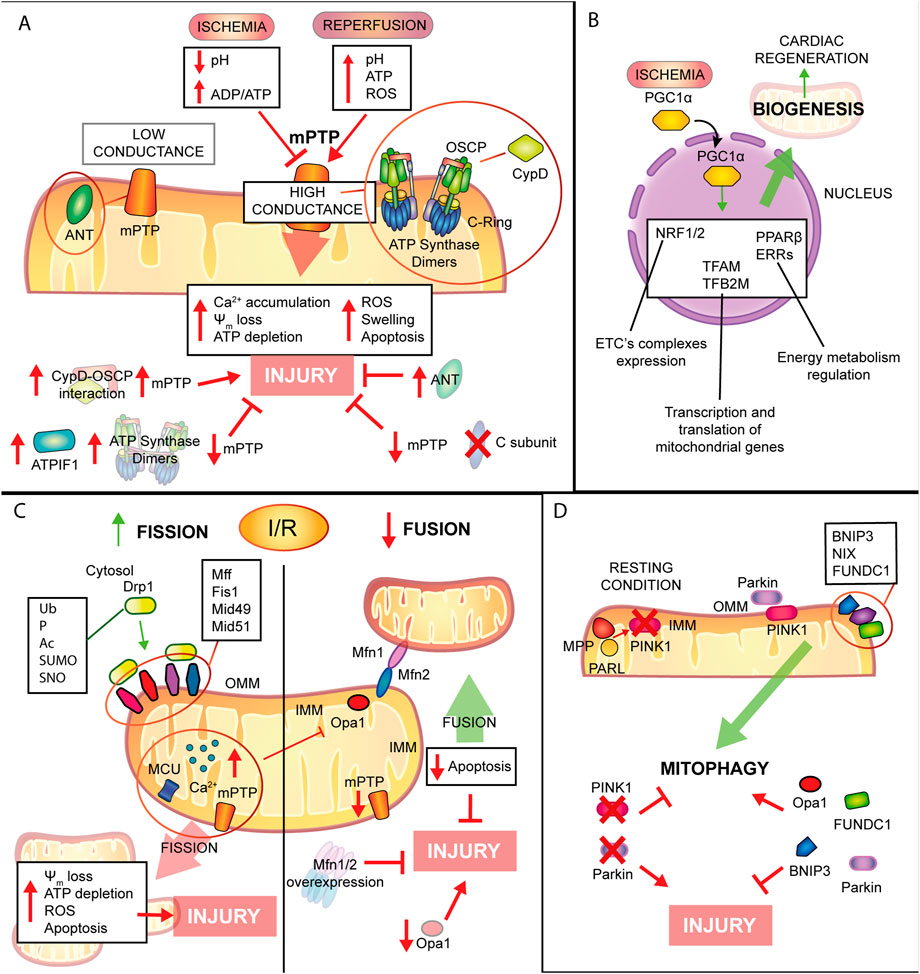
FIGURE 4. Mitochondrial remodelling and state of health in I/R condition. (A) mPTP involvement in IRI. Schematic representation of the main molecular events involving mPTP in IRI. During ischemia mPTP opening is inhibited, after reperfusion this multiprotein complex opening triggers mitochondrial reactions which lead to cardiomyocytes death. (B). Mitochondrial biogenesis. The biogenic process is activated during ischemia with positive consequences in order to improve cellular metabolism and energy production and to promote cardiac regeneration. (C). Mitochondrial dynamics. Fission process is enhanced during I/R leading to tissue injury. Contrariwise, mitochondrial fusion is reduced and increase of this process showed beneficial effects against IRI. (D). Mitophagic process. Induction of mitophagy during myocardial infarction has cardioprotective effects on the condition that is not a chronic activation, very harmful for cardiac tissue.
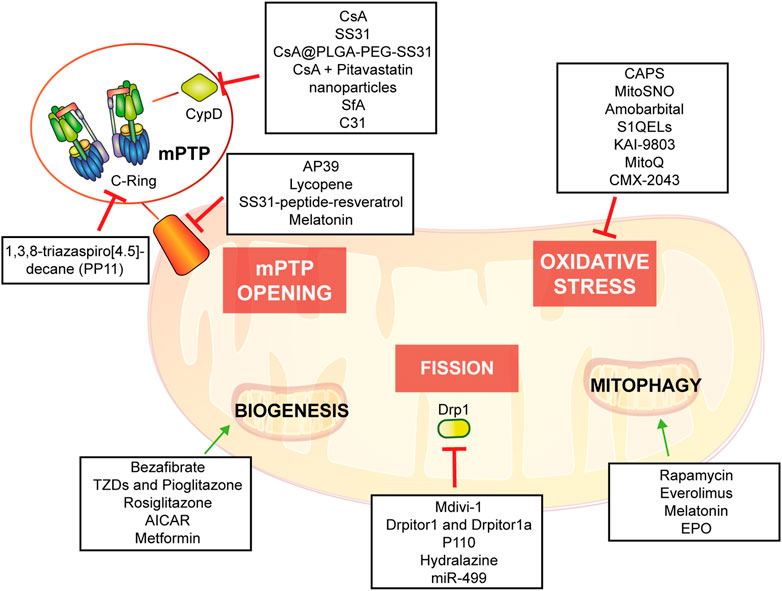
The objective of this review is to describe the relevance of mitochondria in molecular and cellular mechanisms underlying IRI and to describe up-to-date evidence of mitochondria-based potential therapies providing an in-depth understanding of possible therapeutic intervention (Figure 5).
FIGURE 5. Potential mitochondria-targeting therapied as new approach to control heart tissue damage.
Inter-organelle communication in I/RMitochondria-endoplasmic reticulumA physical and functional crosstalk between mitochondria and other organelles, such as plasma membrane (PM), nucleus and sarcoplasmic reticulum (SR) has been previously reported. SR-mitochondria interaction, called mitochondria-associate ER/SR membranes (MAMs), is the first and the most described contact, and it involves several proteins that guarantee a correct tethering and communication between the two organelles (Pinton, 2018; Missiroli et al., 2017; Marchi et al., 2017; Pedriali et al., 2017). In cardiac muscle, SR-mitochondrial contact is a critical regulator of Ca2+ cycle during excitation-contraction coupling (ECC), a process that requires a huge amount of ATP supplied by mitochondria during every heartbeat (Denton and McCormack, 1990; Bers, 2008; Cao et al., 2019). Briefly, upon action potential, Ca2+ enters from the extracellular space into the cytosol through L-Type Ca2+ channel located on the PM, stimulating a large Ca2+ release from the SR via type 2 ryanodine receptor (RyR2), in a calcium-induced calcium released (CICR) manner, ultimately promoting cardiomyocyte contraction. Activation of the sarcoendoplasmic reticulum Ca2+ ATPase (SERCA) brings back Ca2+ into the SR, the main intracellular Ca2+ store, together with Na+/Ca2+ exchanger (NCX) on the PM, which clears out Ca2+ from the cytosol, promoting cardiomyocyte relaxation. The ECC occurs at the dyadic clefts, the space between the SR and T-tubule membranes, but mitochondria are excluded from this zone (Sharma et al., 2000). Thus, it is still of interest to answer the question whether and how mitochondria contribute to buffering Ca2+ for energy production on a beat-to-beat basis (O'Rourke and Blatter, 2009; Fabiato, 1983; De la Fuente and Sheu, 2019).
As already described in the previous section, once Ca2+ is released by the SR via RyR2, it diffuses into the mitochondria intermembrane space via voltage-dependent anion channel 2 (VDAC2) and then is taken up into the mitochondrial matrix via mitochondrial calcium uniporter complex (MCUcx) (Giorgi et al., 2018). It has been reported that MCUcx is an ion channel with low affinity for calcium but highly selective (Kirichok et al., 2004). Thus, in order to make possible the mitochondrial Ca2+ uptake, Ca2+ transfer between SR-mitochondria occurs at the microdomain level (Ramesh et al., 1998; Sharma et al., 2000) where the ions channels assigned for SR-Ca2+ release and mitochondria-Ca2+ uptake are closely connected. Early studies using transmission electron microscopy and electron tomography (Boncompagni et al., 2009; Hayashi et al., 2009), have shown the existence of these microdomains, but the proteins involved and the molecular structure of these tethers are still under investigation (Filadi et al., 2018). Mitofusin 2 (Mfn2) is the first protein identified to be part of the tethers between ER and mitochondria, however contrasting findings regarding its role have been reported. As it will be described, Mfn2 is a mitochondrial GTPase protein involved in mitochondrial fusion, which has been found to localized also at the ER/SR, where it can form homo- or heterodimers with either Mfn2 or Mfn1. Chen et al. showed that mitofusin 1 (Mfn1) deletion, during the embryonic stage, reveals no differences in distances and Ca2+ levels in both SR and mitochondria (Chen et al., 2012). On the contrary, cardiomyocytes specific Mfn2 gene deletion induces a “dissociation” between the two organelles, leading to a Ca2+ rise in SR, which is accompanied by a reduction in mitochondrial Ca2+ uptake, and a lower mPTP activation during reperfusion (Papanicolaou et al., 2011), thus protecting the myocardium from IRI.
In agreement with the previous study, Hall et al. performed a cardiac-specific deletion of both Mfn1 and Mfn2, leading to the disruption of the tethering SR-mitochondria, attenuating ROS burst and mitochondrial Ca2+ overload and showing that hearts deficient in both Mfn1 and Mfn2 are less sensitive to IRI (Hall et al., 2016).
Another complex involved in the SR-mitochondria tethering is the VDAC1/GRP75/IP3R1 channel complex. The mitochondrial stress 70 protein, also known as GRP75, is the bridge between the IP3R1, on the ER side, and VDAC1 at the outer mitochondrial membrane (OMM) (Perrone et al., 2020). Interestingly, during hypoxia and reoxygenation, endothelial cells exhibit an increase in the interaction among the proteins of the VDAC1/GRP75/IP3R1 complex, promoting mitochondrial Ca2+ overload and subsequent cell death (He et al., 2015). Moreover, Paillard et al., identified CypD, a Ca2+ sensitive mitochondrial chaperone known as a regulator of mPTP opening, as a new player in the VDAC1/GRP75/IP3R1 complex (Paillard et al., 2013). These authors performed experiments on cardiomyoblasts and cardiomyocytes and, through independent genetic deletion and pharmacological inhibition of each component of the complex under hypoxia-reoxygenation condition, cardiac cells showed a reduction in the mitochondrial Ca2+ uptake and a reduction of the mPTP opening that correlates with disruption of the SR-mitochondria tethers and Ca2+ signaling (Paillard et al., 2013). In addition, these authors obtained the same results upon deletion of Mfn2, confirming its key role in maintaining the correct mitochondrial-SR Ca2+ crosstalk, fundamental for cellular response to IRI (Paillard et al., 2013).
In a more recent study, also glycogen synthase kinase 3β (GSK3β), a serine/threonine-protein kinase for glycogen synthase, has been identified as a regulator of the VDAC1/GRP75/IP3R1 complex (Gomez et al., 2016). It was already reported that phosphorylation of GSK3β reduces mPTP opening mediating cardioprotection (Nishihara et al., 2007; Juhaszova et al., 2004; Gomez et al., 2008; Tong et al., 2002), but the mechanism of action was still unknown. The authors proposed that upon ischemia and reperfusion (I/R), GSK3β localizes at the MAMs and physically interacts with each protein of the Ca2+-channel complex (VDAC1/GRP75/IP3R1/CypD) and not with RyRs, suggesting a new role as a structural protein of the complex (Gomez et al., 2016). Accordingly, pharmacological inhibition of GSK3β promotes myocardial protection by reducing mitochondrial Ca2+ uptake, which is due to disruption of the interaction of IP3R with the other proteins forming the complex, and not due to the binding of GSK3β on IP3R, which remains unchanged (Gomez et al., 2016).
Another less studied protein is the mitochondrial protein tyrosine phosphatase interacting protein 51 (PTPIP51) which is known to interact with the ER vesicle-associated membrane protein-associated protein B (VAPB) forming a bridge between the two organelles and regulating Ca2+ homeostasis (De Vos et al., 2012). Qiao et al. showed that PTPIP51 is upregulated upon I/R, leading to cardiomyocytes cell death (Qiao et al., 2017). During myocardial infarction, PTPIP51 promotes mitochondria-SR contact, therefore enhancing Ca2+ transfer from SR to mitochondria.
The mitophagic protein fun14 domain-containing protein 1 (FUNDC1) is known to localize at the OMM where it interacts with microtubule-associated protein 1A/1B-light chain 3 (LC3) for autophagosome recruitment; interestingly, it has been recently shown a key role of FUNDC1 as an ER-mitochondrial tethers (Wu et al., 2017; Wu et al., 2016). During hypoxia FUNDC1 interacts with calnexin, an ER protein, and promotes mitochondrial fission by recruiting dynamin-related peptide 1 (Drp1) (Wu et al., 2016). Moreover, another group reported that FUNDC1 modulates ER-Ca2+ release by binding IP3R2. Indeed, ablation of FUNDC1 induces ER-mitochondrial dissociation, decreasing mitochondrial Ca2+ uptake, promoting mitochondrial fission and cardiac dysfunction upon ischemic stress (Wu et al., 2017).
Other two proteins, known as regulators of apoptosis, were identified in the ER-mitochondrial communication: the mitochondrial fission protein 1 (Fis1) and B cell receptor activators (BAP31) at the ER membrane (Iwasawa et al., 2011). During hypoxia-reoxygenation Fis1 stimulates SR-Ca2+ release through interaction with BAP31, which, in turn, promotes Bax and Bak translocation to the mitochondria and the initiation of apoptosis (Lu et al., 2010). In agreement with these studies, a recent article by Cheng et al., has shown that loss of the Fis-BAP31 crosstalk protects hearts from myocardial infarction by downregulating mitochondrial ROS production and c-Jun N-terminal kinase (JNK) pathway activation, thus inhibiting cardiomyocyte cell death (Cheng et al., 2021).
All together, these data highlight the importance of the correct interaction between SR-mitochondria for the maintaining a correct Ca2+ homeostasis: a reduction in the ER-mitochondria tethering correlates with a decrease in mitochondrial Ca2+ uptake, reducing cardiac myocytes cell death, thus providing a novel platform for preventing IRI.
Mitochondria-plasma membraneThe contact between the mitochondria and the PM has been disregarded and, to our knowledge, only few insights delineated mitochondria-PM relationships and their molecular elements. This contact might have an extensive impact on mitochondrial function and cell physiology, but results of these studies are still limited and the molecular bases of this crosstalk are still unclear. Multiple observational studies over the past 50 years have proposed a specific attachment between the mitochondria and PM in neuronal and epithelial cells using electron microscopy, in which the mitochondria-PM contact has been found in close proximity (Fridolfsson et al., 2012; Montes de Oca Balderas, 2021).
The mitochondria-PM interaction has been poorly described at the molecular level in yeast (Westermann, 2015). During yeast cell division mitochondria exhibit a tether, termed the mitochondria-ER cortex anchor, with the PM which is mainly mediated through nuclear migration 1 (Num1) and mitochondrial distribution and morphology 36 (Mdm36). Num1 plays an essential role in the mitochondria-PM interaction in which it anchors directly with the OMM to PM by its pleckstrin homology domain in PM, as well as to the mitochondrial network through the coiled-coil domain, where cardiolipin, in addition to other proteins negatively charged located to the cristae junction, is mainly involved (Ping et al., 2016). Moreover, Num1 forms a complex with Mdm36 and their localization is on the cortical part of the mitochondrial tubules (Lackner et al., 2013); it has been shown that this complex has significant effects on mitochondrial inheritance and dynamics via the mitochondria-PM tethering (Lawrence and Mandato, 2013). Num1/Mdm36 complex plays an essential role in the mitochondrial morphology and both proteins are considered as key factors of a mitochondria-PM tether complex (Westermann, 2015). Therefore, it has been suggested that other proteins participate to the mitochondria-PM contact beyond the complex Num1/Mdm36 (Lackner et al., 2013), even if more in-depth analyses are needed. Further research should focus on the identification of new proteins orchestrating these processes and their effect on the contact sites.
In cardiomyocytes from multiple species, one piece of evidence has identified a close cohesion of mitochondria to the gap junctions (GJs) of the PM (Forbes and Sperelakis, 1982). The decrease of cell coupling through inhibition of GJs during reperfusion leads to reduced IRI (Rodriguez-Sinovas et al., 2004). In addition, the involvement of GJs has been intensely studied in ischemic preconditioning-induced protection against myocardial reperfusion injury (Miura et al., 2010).
Experimental studies showed that caveolae (PM invaginations which contribute to cell response) are present in cardiomyocyte’s T-tubules located near the mitochondria at the PM (Burton et al., 2017). Consistently, caveolae were also investigated in other studies and found in the cardiomyocytes near the SSM, disclosing a crucial impact on the cell physiology (Fridolfsson et al., 2012). Intriguingly, the mitochondria-associated caveolae number augmented in response to I/R indicating a dynamic relationship linking the mitochondria and the PM (Fridolfsson et al., 2012; Foster et al., 2020). In support of this hypothesis, Fridolfsson and others studied the translocation of caveolin-3 (Cav-3) from the PM to the inner mitochondrial membrane (IMM) in response to cellular stress (Fridolfsson et al., 2012). In the same study, it was shown that Cav-3 overexpression in mouse models provides protection from IRI: cardiac mitochondria exhibit an increase in respiration, improved Ca2+ tolerance to higher concentrations and reduced ROS generation. Mice overexpressing Cav-3 showed a reduced infarct size, whereas Cav-3 deletion increased mitochondrial dysfunction. These data suggest a cardioprotective effect of Cav-3 against I/R linked to cell protection and cell adaptation to stress (Fridolfsson et al., 2012). Other studies reported that Cav-1 deletion might also play a protective role in vivo I/R mouse models (Tsutsumi et al., 2010). In conclusion, the above described evidences on the involvement of Cav-3 and caveolae in stress response to I/R, might represent an innovative way to counteract tissue damage. Anyway, additional experimental studies are required to unveil the key roles of mitochondria-PM during I/R in mammals.
Mitochondria-nucleusAs previously described, cardiomyocytes possess different types of mitochondria. Concerning PNM, these mitochondria have different morphology, reduced Ca2+ uptake, exhibit increased fusion and fission activity compared to other pools, suggesting different roles among them (Lu et al., 2019). In fact, it has been shown that mitochondrial position inside the cell can control specific Ca2+ signaling through a local regulation (Park et al., 2001). Besides Ca2+, the perinuclear localization of mitochondria can control nuclear transport across the nuclear envelope, optimizing the distance of energy transfer. Indeed, it ensures the energetic supply necessary for the intense crosstalk nucleus-cytoplasm (Dzeja et al., 2002). The mitochondrial subpopulations are differently associated with different pathological condition. For example it is known that, in the IRI process, OXPHOS is decreased only in SSM type and not IFM one, suggesting the importance of mitochondrial heterogeneity in the pathophysiology of this condition (Lesnefsky et al., 1997).
Mitochondria-nucleus connection is fundamental for the stability of mitochondria: almost all the mitochondrial proteins are encoded by the nucleus and also biogenesis is finely regulated both by mitochondrial DNA (mtDNA) and nuclear DNA (Garesse and Vallejo, 2001). The nucleus controls mitochondrial stability through anterograde signals. Molecular components of this finely regulated system include a number of nuclear genes encoding mitochondrial proteins controlled by different intra- and extra-cellular signals (Ng et al., 2014), in addition, it has been deeply studied the involvement of microRNAs (miRNAs) in the regulation of the nucleus-mitochondria communication. In particular, cardiac tissue is characterized by miR-30 family members (Ikeda et al., 2007) and they have been associated to apoptotic process through regulation of mitochondrial fission machinery by targeting p53-Drp1 axis. miR-30 targets p53 inhibiting Drp1 transcription, thus blocking both mitochondrial fission and apoptosis (Li et al., 2010). Recurring proofs have connected altered miRNA expression to myocardial IRI. Thanks to animal model of I/R and tools of overexpression or inhibition of specific miRNA, it has been defined the opposite role of miRNAs in IRI pathogenesis, or rather, pro-apoptotic or anti-apoptotic acting on different pathways (Yang et al., 2014; Zhu et al., 2018). For instance, in vitro experiments in cardiomyocytes have confirmed a role of miR-378 in controlling the level of cell death during ischemia and this miRNA repressed caspase-3 expression reducing both apoptosis and necrosis, thus paving the way to new potential therapies based on miRNA targeting (Fang et al., 2012). A recent study on mice model of I/R has linked miR-138 to hypoxia-inducible factor 1α (HIF-1α). This miRNA showed a cardioprotective role in reducing infarct size and myocardial enzymes, inhibiting expression of cleaved caspase-9, caspase-3 and of HIF-1α (Liu et al., 2019). Another example is given by in vitro studies on cardiomyocytes linking increased cardiac damage to the decrease of miR-146a levels after I/R: in normoxic conditions miR-146a localizes to the mitochondria where it modulates CypD protein expression, it prevents mPTP opening and the apoptotic cascade (Su et al., 2021). On the contrary, through knockdown approaches, nuclear miR-762 increase has been associated to apoptosis consequent to I/R: after the treatment, this miRNA translocates to mitochondria, where it interacts with mitochondrial NADH dehydrogenase subunit 2 (ND2) modulating ATP and ROS production and the consequent cardiomyocytes apoptosis (Yan et al., 2019).
It is known that dysfunctions in this inter-organelle communication leads to serious damage to the intricate balance of cellular homeostasis. Indeed, inducers of mitochondrial retrograde signaling such as Ca2+-dependent stress, mtDNA damage, metabolical disorders and loss of mitochondrial membrane potential (ψm) permit to impaired mitochondria to communicate with nucleus as a compensative mechanism (Guha and Avadhani, 2013). Recently, new contact sites have been identified: NAM (Nucleus-associated mitochondria) are points of contact of mitochondria with nucleus to favor communication. This retrograde type of communication is mediated by cholesterol, ROS and Ca2+ and the tethering between the two organelles is permitted by a complex of different protein such as translocator protein (TSPO), protein kinase A (PKA), A-kinase anchoring protein acyl-CoA binding domain containing 3 (ACBD3), but others might be involved (Desai et al., 2020).
As already explained, one of the main mitochondrial functions is the control of apoptosis through the release of pro-apoptotic factors such cytochrome c and activation of caspase-3 and -9, a mechanism that has been shown to be activated also during reoxygenation after hypoxia in vitro experiments on adult cardiomyocytes (Kang et al., 2000). Another mitochondrial effector of apoptotic cell death, independent from the caspases, is Apoptosis Inducing Factor (AIF): it is released from the mitochondria and translocates to the nucleus where it induces chromatin condensation (Susin et al., 1999). The apoptotic role of AIF has been confirmed also in the IRI process. In vivo mice hearts subjected to occlusion of the coronary artery, AIF release and nuclear translocation have been considered as a readout of tissue damage. Of interest is the observation that in mice knockout for Hsp70, I/R-induced cardiac dysfunction is amplified, probably because Hsp70 inhibits AIF translocation by direct interaction (Choudhury et al., 2011).
Another type of mitochondria-nucleus communication is through a nuclear cofactor, peroxisome proliferator activated receptor gamma coactivator 1-alpha (PGC-1α), which interacts with both transcription and nuclear factors controlling the relationship between the two organelles. In the IRI condition, PGC-1α is activated by increased ROS and Ca2+ and in turn stimulates several mitochondrial related genes, linked to antioxidant system or mitophagy process with a general effect of cardioprotection (Li Y. Q. et al., 2022).
In conclusion, even if the evidences concerning this inter-organellar communication are still limited and more studies are needed, several findings highlighted its importance in the pathophysiology of IRI and in the overall homeostasis of cardiomyocytes.
Mitochondrial remodeling and state of health in I/R conditionFission-fusionMitochondria are highly dynamic organelles able to change their number, morphology, and distribution (Tilokani et al., 2018). These changes are coordinated by two crucial events: fission and fusion that define the mitochondrial dynamics in response to the changes in the metabolic status of cardiomyocytes as well as environmental variations. Both processes are pivotal for maintaining mitochondrial integrity and functions, cell cycle modulation and cell quality control (Liu et al., 2020), counterbalancing each other, they are strictly orchestrated by core protein machinery that are mainly large guanosine triphosphates GTPases that exhibit membrane-remodeling characteristics (Lee and Yoon, 2016).
In mammals, mitochondrial fission is coordinated by GTPase Drp1, mitochondrial fission factor (Mff), Fis1, and 49kD and 51kD mitochondrial dynamics proteins (Mid49/51) (Tong et al., 2020). The initiation of mitochondrial fission is mediated by a stringent replication of the mtDNA in the matrix (Lewis et al., 2016). In physiological conditions, mitochondrial fission is relatively less compared with stress conditions, in which Drp1 undergoes post-translational changes including ubiquitination, phosphorylation, acetylation, SUMOylation, and S-nitrosylation (Hu et al., 2020). When activated, Drp1, the master regulator of mitochondrial fission, actively translocates from the cytosol to the OMM surface, where it interacts and binds to its non-GTPases receptors such as Fis1, Mff, and Mid49/51 (Hall et al., 2014).
Mitochondrial fission serves to meet high energy demands: it cleaves a mitochondrion into two daughter mitochondria and it enables clearing the damaged mitochondria from the cardiomyocytes, maintaining the separation of damaged mitochondrial fractions from the whole network. This process is recognized as a prerequisite for selective mitochondrial autophagy termed “mitophagy”, by which damaged mitochondria containing altered proteins, disrupted mtDNA or membranes, are cleared (Suen et al., 2010).
Mitochondrial fusion is regulated by three GTPase dynamin proteins including Mfn1 and Mfn2 and optic atrophy factor 1 (Opa1). The two mitofusins are anchored to the OMM and form homotypic or heterotypic oligodimers to cooperate or work individually to activate OMM fusion with adjacent mitochondria. In contrast, Opa1 is an IMM protein that induces IMM intermingling through the cristae structure regulation (Cohen and Tareste, 2018). Several efforts have been made to define the exact structure and conformational changes of Mfns occurring after phosphorylation, for example through comparative analysis (Mattie et al., 2018) and generation of small-molecule agonists it has been described the specific effect of PINK1 in modulation Mfn2 activity (Rocha et al., 2018).
This dynamic process of fusion consists of two neighboring mitochondria’s OMM and IMM that fuse and share material: this is central for mitochondrial health and physiological functions, including maintaining the balance of matrix metabolites, membrane components and an intact mtDNA (Hoppins et al., 2007).
Mitochondrial remodeling during myocardial I/R involves structural and metabolic changes, both of which are known to play an essential role in each stage of cardiac I/R pathogenesis. During normal physiological conditions, balanced mitochondrial fission with fusion provides an initial step in the culling of defective organelles from the mitochondrial network and facilitates the mitochondrial trafficking to specific subdomains in the membrane inside the cell termed microdomains (Hom and Sheu, 2009; Chang and Reynolds, 2006).
Mitochondrial fission has been shown to be upregulated both during ischemia and reperfusion, when it ultimately decreases ATP production efficiency and disrupts the ψm, thus leading to cardiomyocyte apoptosis (Maneechote et al., 2017). Conversely to mitochondrial fission, when the mitochondrial fusion is downregulated, it promotes mitochondrial fragmentation and rises cardiac cell death in response to I/R (Hall et al., 2016). For example, Bradly and others firstly identified extensive mitochondrial fragmentation during myocardial ischemia in HL-1 cells (Brady et al., 2006). Mitochondrial segregation splits the elongated mitochondrial network into short rods, activating mitochondrial depolarization, ATP depletion, and upregulating ROS generation and apoptosis (Hall et al., 2016). These findings showed that the repression of fission machinery prevents the mitochondrial fragmentation and swelling due to Ca2+ overload and were confirmed in various I/R models, both in vitro, ex vivo and in vivo. The increase in mitochondrial fusion reduces the susceptibility to mPTP opening and to cardiomyocytes apoptosis, leading to amelioration of the diastolic, contractile function and reduction in infarct size (Ong et al., 2010; Sharp et al., 2014).
Promoting mitochondrial fusion and repressing excessive mitochondrial fission have been proposed as promising protective mechanisms against myocardial damage after I/R, even if there are some conflicting results in the literature. Indeed, the overexpression of fusion proteins Mfn1 or Mfn2 protects HL-1 cells from IRI through the promotion of mitochondrial fusion (Hall et al., 2016). Also, mitofusins overexpression with the repression of Drp1-mediated fission rescues the cardiomyocytes against the mPTP opening, which is a critical determinant of the cell death process during IRI (Hausenloy and Yellon, 2003; Ong et al., 2010). However, mitofusins deletion increases mitochondrial segregation, thus stimulating the mitochondrial apoptosis in response to I/R (Griparic et al., 2004). Another study, instead, showed that the cardiomyocyte-specific dual deletion of mitofusins reduces cell death after acute myocardial I/R by blocking mPTP activity, thus preventing the mitochondria from elevated Ca2+ concentration (Hall et al., 2016). More studies are needed in order to understand the contribution of fission and fusion processes to IRI.
Of note, enhanced expression of mitochondrial calcium uniport (MCU) significantly upregulates Ca2+ levels, in return inducing the opening of mPTP and leading to the elevation of calpain expression, which has been shown to inhibit Opa1 expression and to stimulate calcineurin, causing Drp1 phosphorylation and excessive fission in mice models of hypoxia/reoxygenation and myocardial I/R (Patergnani et al., 2020; Guan et al., 2019).
During hypoxia, HIF-1α is a central regulator mediating the cellular response and plays a key role in multiple CVDs, such as IHD (Bouhamida et al., 2022). Interestingly, the expression of the mitochondrial-targeted HIF-1α significantly decreases Drp1 mitochondrial association and Opa1 levels, as well as attenuates apoptosis mediated by hypoxia (Li H. S. et al., 2019). Further experimental studies may provide a better understanding of the molecular mechanisms behind the correlation between the mitochondrial fission-fusion and hypoxia in response to I/R.
The mitochondrial fusion protein Opa1 is intimately involved in the remodeling of cristae and in control of the release of cytochrome c, while its reduction, through the elevation of ROS, aggravates cardiomyocyte apoptosis during I/R (Shen et al., 2007). In support of this finding, Opa1 deficiency has been identified to correlate with I/R-mediated mitochondrial segregation (Ong et al., 2010; Chen et al., 2008) and additionally, Chen and others reported decreased Opa1 levels in human hearts samples with ischemic cardiomyopathy (Chen et al., 2008).
Further insights were obtained using melatonin, a potent mitochondrial-targeted antioxidant (Tan et al., 2016) that induces Opa1 expression at transcriptional level via the AMP-activated protein kinase (AMPK) signaling pathway and it increases the mitophagic process, leading to a cardioprotective effect against the IRI (Zhang et al., 2019c).
Pioneering studies have documented the important role of Mff in mediating fatal mitochondrial fission in response to I/R. For instance, Zhou et al. have reported that enhanced levels of nuclear receptor subfamily four group A member 1 (NR4A1) increases, in return, serine/threonine kinase casein kinase two α (CK2α), which promotes Mff mediated-excessive fatal mitochondrial fission, leading to an impaired mitochondrial function and subsequent activation of apoptosis during IRI (Zhou et al., 2018a). In the light of these findings, the higher levels of Mff induced by JNK cause an excessive mitochondrial fission and the activation of mitophagy, thereby triggering cardiomyocyte death (Jin et al., 2018). The phosphorylated Mff elevates the translocation of the cytoplasmic Drp1 into the mitochondria, and the interaction between Mff and Drp1 stimulates mitochondrial segregation, resulting in excessive ROS production, the dissociation of hexokinase 2 (HK2), mPTP opening, and subsequent stimulation of the apoptotic process during cardiac IRI (Zhou et al., 2017).
In addition to the findings mentioned above, Zhang et al. identified that the repression of Socs6, a cyclic AMP-dependent kinase phosphorylation site that inhibits the Drp1 translocation to the mitochondria through the QK/miR-19b/Socs6 signaling pathway attenuates hypoxia/reperfusion-mediated mitochondrial fission and cardiomyocyte impairment in vitro and in vivo mouse models (Zhang et al., 2022).
Taken together, these findings suggest a major role of mitochondrial fission and fusion in response to myocardial stress such as what occurs during I/R. Thus, the stimulation of mitochondrial fusion factors may have a promising impact on the prevention of myocardial damage during this condition. In spite of the availability of a number of advanced studies focused on the understanding the mitochondrial dynamics, there are only relatively few data correlating I/R in animal models in the regulation of this process. Thus, further insights concerning this issue may provide new therapeutic interventions that are beneficial in cardioprotection.
Mitochondrial turnover: A balance between biogenesis and mitophagy in IRIAs already mentioned, cell homeostasis requires a fine balance among the three mitochondrial quality control processes: mitochondrial dynamics, already discussed in the previous chapter, biogenesis and mitophagy. Mitophagy is a specific, evolutionary conserved, subtype of macroautophagy, which promotes physiological degradation of senescent or irreversibly damaged mitochondria (Morciano et al., 2020). Therefore, impairment in the mitophagic process, leads to tissue damage, due to accumulation of defective organelles (Morciano et al., 2021a). Canonical mitophagy is controlled by PINK1/Parkin pathway: in summary, under resting condition PTEN-induced putative kinase1 (PINK1) is transported into the IMM where it is degraded by mitochondrial proteases matrix processing peptidase (MPP) and presenilis-associated rhomboid-like protein (PARL) (Jin et al., 2010). Upon loss of ψm, PINK1 accumulates at the OMM to directly recruit Parkin RBR E3 ubiquitin-protein ligase (Parkin) from the cytosol (Koyano et al., 2014), or indirectly by phosphorylation of Mfn2 and promoting Mfn2 Parkin-mediated ubiquitination (Chen and Dorn, 2013). Recent discoveries by Li et al. demonstrate that PINK1-mediated phosphorylation is essential and sufficient, to switch Mfn2 from fusion protein, when not phosphorylated, to mitophagy promoter after phosphorylation on T111, S378 or S442 and that Parkin is not necessary for negative regulation of Mfn2-mediated mitochondrial fusion, but involved in Mfn2 control of mitophagy (Li J. et al., 2022).
Parkin phosphorylation at Ser65 increases its E3 ligase activity, thus promoting poly-ubiquitination of mitochondrial proteins and phagosome recruitment for subsequent protein degradation by the lysosome (Koyano and Matsuda, 2015; Sciarretta et al., 2018). Of note, mutations in the PINK1/Parkin pathway were firstly associated with autosomal recessive Parkinson’s disease (Kitada et al., 1998), but, of interest, extensive studies in cardiovascular diseases highlighted that impairment in PINK1/Parkin pathway promotes alterations in the mitophagic flux leading to cardiac dysfunction. Indeed, mice lacking PINK1 develop cardiac hypertrophy (Billia et al., 2011) and are more susceptible to IRI (Siddall et al., 2013), as well as are associated with cardiac mitochondrial dysfunction and increased ROS production (Billia et al., 2011; Siddall et al., 2013). Moreover, another study on Parkin-deficient mice reported a reduction of mitophagic markers in the border zone of the infarct associated with accumulation of dysfunctional mitochondria (Kubli et al., 2013).
It should be noted that there is a crosstalk among all the mitochondrial quality control mechanisms. For instance, mitochondrial fission is commonly considered a pre-step of mitophagy activation. Downregulation of Drp1 induces, not only mitochondrial fusion, but also an increase of damaged mitochondria due to inhibition of mitophagy, thus worsening myocardial infarction (Ikeda et al., 2015; Song et al., 2015). On the contrary, it has been shown that Opa-1-induced mitophagy mediated cardioprotection against hypoxia-induced apoptosis (Xin and Lu, 2020), and that AMPK-Opa1 signaling pathway induced mitochondrial fusion/mitophagy upon melatonin treatment thus attenuating cardiomyocyte death and mitochondrial stress in the setting of cardiac IRI (Zhang et al., 2019c). According to that, cardiac myocytes overexpressing Parkin, are more protected against hypoxia-mediated cell death. These data suggest that a mild induction of mitophagy in myocardial infarction has a beneficial role to clear dysfunctional mitochondria, whereas chronic activation of mitophagy may be detrimental leading to chronic heart diseases such as hypertrophy and heart failure (Shires and Gustafsson, 2015; Ramaccini et al., 2020; Morciano et al., 2020).
In addition, mitophagy may also occur through the ubiquitin-independent mechanism, mediated by adaptor proteins located at the OMM which contain an LC3 interacting motif, thereby recruiting autophagosomes to damaged mitochondria. These receptors include: BNIP3 (BCL2/Adenovirus E1B 19 KDa Protein-Interacting Protein 3), BNIP3L, also known as NIX, (BCL2 interacting protein three like) (Zhang and Ney, 2009; Zhang and Ney, 2011; Lee et al., 2011) and FUNDC1 (Liu et al., 2012). Several studies showed the role of these proteins in acute I/R by modulating mitophagy, but the role of BNIP3 and NIX in the heart is still unclear, since these two proteins exhibit both the capacity to induce cell death and to promote autophagy (Zhang and Ney, 2009).
It has been reported that upon hypoxia, mitophagy is upregulated through activation of the HIF-1α/BNIP3 axis, promoting cardioprotection by removing damaged mitochondria and promoting myocardial remodeling after IRI (Zhang et al., 2008; Bouhamida et al., 2022; Hamacher-Brady et al., 2007; Zhang et al., 2019b). However, other studies suggested that after cardiac stress, overexpression of BNIP3 induces cardiomyocytes death and a decreased myocardial function (Regula et al., 2002; Diwan et al., 2007). With regard to the autophagic cell death role, it is unclear whether death is due to excessive autophagy or to an independent death-inducing function of BNIP3 or NIX. FUNDC1 interacts with LC3 upon hypoxia in ischemic heart, promoting mitophagy, which is associated with inhibition of apoptotic cell death; however, after reperfusion, FUNDC1-mediated mitophagy is inactivated, resulting in a boost of cell death (Liu et al., 2012; Zhou et al., 2018b).
Dysfunctional mitochondria are cleared by mitophagy, whereas mitochondrial population and mass are maintained by activation of mitochondrial biogenesis (Palikaras et al., 2015). Mitochondria cannot be synthesized ex novo, and in resting conditions, mitochondrial turnover in the adult heart occurs every 2 weeks (Dorn et al., 2015). Moreover, mitochondrial biogenesis is activated and increased in response to high energy demands, such as right after exercise or cold, or under stress conditions, such as hypoxia and oxidative stress (Ham and Raju, 2017). Mitochondria are unique and semiautonomous organelle, which contain their own mtDNA. However, mtDNA encodes only 23 essential subunits of the electron transport chain (ETC), as well as all rRNAs and tRNAs. Therefore, mitochondrial biogenesis requires a fine-tuned coordination of four processes: transcriptional activation of nuclear-encoded mitochondrial genes, mitochondrial protein translocation, replication of mtDNA and synthesis of mitochondrial phospholipids (Sun et al., 1990; Dorn et al., 2015).
To date, the mechanisms controlling the mitochondrial biogenesis process are not fully understood. Several transcriptional factors are involved in this process and PGC-1α is considered the master regulator of mitochondrial biogenesis with a key role also in oxidative metabolism and antioxidant defenses in mammalian cells (Fernandez-Marcos and Auwerx, 2011). Briefly, during mitochondrial biogenesis, PGC-1α translocates into the nucleus where it co-activates nuclear respiratory factors 1 and 2 (NRF1 and NRF2), which are regulators of the expression of subunit of ETC’s complexes, or mitochondrial transcription factor A (TFAM) and mitochondrial transcription factor B2 (TFB2M), required for transcription and translation of mitochondrial genes (Gleyzer et al., 2005; Scarpulla, 2008; Ding et al., 2021). Other transcriptional factors that are activated by PGC-1α are peroxisome proliferator-activated receptor (PPARβ) and estrogen receptor-related receptor-alpha (ERRs), which are mainly involved in energy metabolism regulation (Giguere, 2008).
Several studies have reported downregulation of PGC-1α expression in the failing heart, which is commonly associated with low ATP production and reduced energy metabolism (Pisano et al., 2016; Riehle et al., 2011). Mitochondrial biogenesis is also reduced in cardiac IRI: recent studies reported that PGC-1α is induced by hypoxia as an adaptive mechanism that increases cardiac regeneration, thus facilitating the recovery of infarcted heart (Sun et al., 2007; Honda et al., 2008; Yue Tl et al., 2001). On the same line, Andres et al. propose the detection of PGC-1α in blood samples as a prognostic factor during myocardial infarction (Fabregat-Andres et al., 2011; Fabregat-Andres et al., 2016). They reported that peripheral levels of PGC-1α correlate with expression in cardiac muscle, which are inversely related to cardiac recovery after IRI (Fabregat-Andres et al., 2011; Fabregat-Andres et al., 2016; Fabregat-Andres et al., 2015). Therefore, PGC-1α might represent an interesting target due to its cardioprotective role in myocardial infarction.
An interesting study reports that melatonin is capable to induce AMPK activation (Qi and Wang, 2020), which simultaneously inhibits mammalian target of rapamycin (mTOR) and induces PGC-1α activation, stimulating mitophagy and mitochondrial biogenesis respectively (Xu et al., 2012; Kupr and Handschin, 2015), ultimately protecting the injured heart.
It is interesting to note that the Hippo pathway, well studied in other pathological conditions but just lately come to light into the cardiovascular field, has been found to be less active in cardiac regeneration, while upstream kinases of the pathway are elevated in myocardial infarction (Ramaccini et al., 2022). According to that, a recent work by Chen et al., showed that the deletion of large tumor suppressor kinase 2 (LATS2), an upstream kinases of YAP1, increased cardiomyocyte viability and mitochondrial biogenesis in a PGC-1α-dependent manner, by increasing expression levels of PGC-1α, in association to the rise of mRNA levels of TFAM and NRF1 in cardiomyocytes (Chen et al., 2021). In this scenario, LATS2 suppression ameliorates mitochondrial functions and attenuates myocardial infarction.
In conclusion, both mitophagic process and biogenesis are fundamental for mitochondrial health and general cellular homeostasis, and stimulation of these mechanisms might become a cardioprotective strategy to challenge IRI.
mPTP: A leading role in IRIOf great interest to all the scientific community, the molecular composition of the mPTP has been explored widely over the past 40 years, and different hypotheses have been proposed and retracted during the years. The purpose of this review is not to gather the most recent findings on mPTP structure, but to focus on the contribution of this multiprotein complex in IRI.
In summary, mitochondrial permeability transition induces the opening of two types of pores characterized by different conductance: a low conductance pore that is associated to adenine nucleotide translocator (ANT) (Neginskaya et al., 2019; Karch et al., 2019; Carrer et al., 2021); a high conductance pore linked to F1FO-ATP synthase and its dimer formation or disassembly (Bonora et al., 2017), or the conformational changes of its c-ring (Bonora et al., 2013). These two pores work in balance and this is confirmed by the direct interaction of ANT and F1FO-ATP synthase: the low conductance state is a transitory opening involved in Ca2+ homeostasis, whereas the high conductance state is long-lasting and leads to cell death (Wacquier et al., 2020). An important regulator of mPTP is CypD (a peptidyl-prolyl cis-trans isomerase) (Halestrap et al., 1997), which interacts with oligomycin sensitivity conferral protein (OSCP) of F1FO-ATP synthase (Giorgio et al., 2017): this is confirmed by the most known inhibitor of mPTP, cyclosporine A (CsA), which targets cyclophilin D (CypD). In addition to CypD, several regulators of mPTP have been recognized to be positive or negative actors in this process, either through interaction or post-translational modifications (Bonora et al., 2022).
Pathophysiology of ischemia/reperfusion at cellular level has been robustly elucidated by mPTP contribution in conditioning cell damage (Hausenloy et al., 2020; Morciano et al., 2015), since a bunch of paper and reviews have focused on this aspect. The objective of this subchapter is to give an overall point of view of the process with a perspective on recent studies.
The lack of oxygen after the ischemic event leads to cardiomyocyte dysfunction linked to mitochondrial ability: the ETC reduces its activity and the production of ATP, essential for all metabolic processes (Hausenloy and Yellon, 2013). To counteract energy shortage, cells switch to anaerobic glycolysis causing the rise of lactate and hydrogen ions and consequent cellular acidosis. In healthy conditions, the reestablishment of pH is carried out by PM Na+/H+ exchanger and the consequent accumulation of cytosolic Na+ is compensated by Na+/K+ ATPase, which uses cellular ATP reserves: lack of energy, in the form of ATP, blocks the activity of, not only Na+/K+ ATPase, but also plasma membrane calcium ATPase (PMCA) and SERCA, and the final outcome is an overload of cytosolic Ca2+ (Piper et al., 2003; Bonora et al., 2019). Of note, during ischemia mPTP is closed, inhibited by an increase in the ADP/ATP ratio and by low pH. It is known that the opening of mPTP is optimal at pH 7.4 (Haworth and Hunter, 1979), whereas at pH 6.5 the channel is blocked due to the protonation of the highly conserved His112 of OSCP subunit of F1FO-ATP synthase (Antoniel et al., 2018). Only after few minutes of reperfusion, oxygen recover allows restoration of mitochondrial respiration, ATP production and pH neutralization but also ROS production (Granger and Kvietys, 2015), which stimulates mPTP opening and Ca2+ accumulation in mitochondria (Griffiths and Halestrap, 1995). As already mentioned, the opening of this mitochondrial pore leads to ψm dissipation, ATP production arrest, mitochondrial swelling and finally activation of regulated cell death process (Halestrap et al., 2004; Ong and Gustafsson, 2012; Bonora et al., 2020).
A very recent publication has linked Mitofilin, an IMM protein with function of control of mitochondrial structure and remodeling, to IRI. In particular, it has been shown that mitofilin binds to CypD through its C-terminal (Baines et al., 2005; Nakagawa et al., 2005; Schinzel et al., 2005). This interaction is reduced during the early moments of reperfusion, specifically, after I/R, Mitofilin-CypD interaction is disrupted and the protein levels decrease with an inversely proportional trend to the degree of myocardial infarct size (Tombo et al., 2020). In addition, the amount of myocardial injury on an in vivo I/R model has been correlated to CypD phosphorylation on its S191 residue, which regulates its ability to control mPTP opening and to interact with OSCP (Hurst et al., 2020).
In the past, ANT was suggested as a hypothetical pore-forming component of the mPTP but this evidence has been later rejected. This protein, instead, is an important regulator of mPTP (Karch and Molkentin, 2014). Indeed, mitochondria extracted from hearts of rats overexpressing ANT1, the main isoform expressed in the heart, showed a strong cardioprotection against IRI in terms of mitochondrial Ca2+ retention capacity and ψm, indexes of mPTP opening and mitochondrial oxygen consumption (Dorner et al., 2021). In a previous study, the same group reported that this ANT1-overexpressing in vivo model was strictly related to augmented cell survival of hypoxic cardiomyocytes and this was probably due to the fact that ANT1 controlled oxidative stress by decreasing ROS generation thus protecting from IRI (Klumpe et al., 2016).
Several studies have connected the c subunit of FO-ATP synthase to mPTP functionality and pore formation (Bonora et al., 2013; Azarashvili et al., 2014; Alavian et al., 2014), with particular involvement in the IRI process. Serum levels of c subunit protein correlate with several surrogate markers of myocardial reperfusion in STEMI patients (Campo et al., 2016), and selective compounds targeting the c subunit inhibit mPTP opening and IRI in animal models, ameliorating cardiac function and reducing apoptosis (Morciano et al., 2018; Fantinati et al., 2022). In addition, a mutation in the glycine-rich domain of c subunit, a highly conserved domain, leads to functional and conformational impairments (Huang and Docampo, 2020; Alavian et al., 2014). In particular, in
留言 (0)