
記住我
To the Editor:
Camptodactyly-arthropathy-coxa vara-pericarditis (CACP) syndrome is a rare autosomal recessive disorder characterized by camptodactyly, noninflammatory arthropathy, progressive coxa vara, and aseptic pericarditis. It is more common in countries with high rates of consanguineous marriage, such as Saudi Arabia, the United Arab Emirates, and Pakistan.1-3 We report on the first Chinese families with CACP syndrome, to our knowledge, including the loci of the genetic mutations, and 2 novel pathogenic variants of the proteoglycan 4 (PRG4).
Ethics approval and informed patient consent were obtained from the patients and their guardians. All procedures performed in studies involving human participants were in accordance with the ethical standards of the authors’ institution. Written informed consent was obtained from the parents of children included in this study.
Four children from 2 unrelated families were clinically diagnosed with CACP syndrome and the associated mutations were confirmed. Patients 1 to 3 are siblings who are homozygous for c.1982_1983delCT, and their parents’ marriage is not consanguineous. Patient 4 carries 2 mutations, c.1006G>A and c. 3412a>G, which had not been previously recorded in the human genome mutation database (Figure).
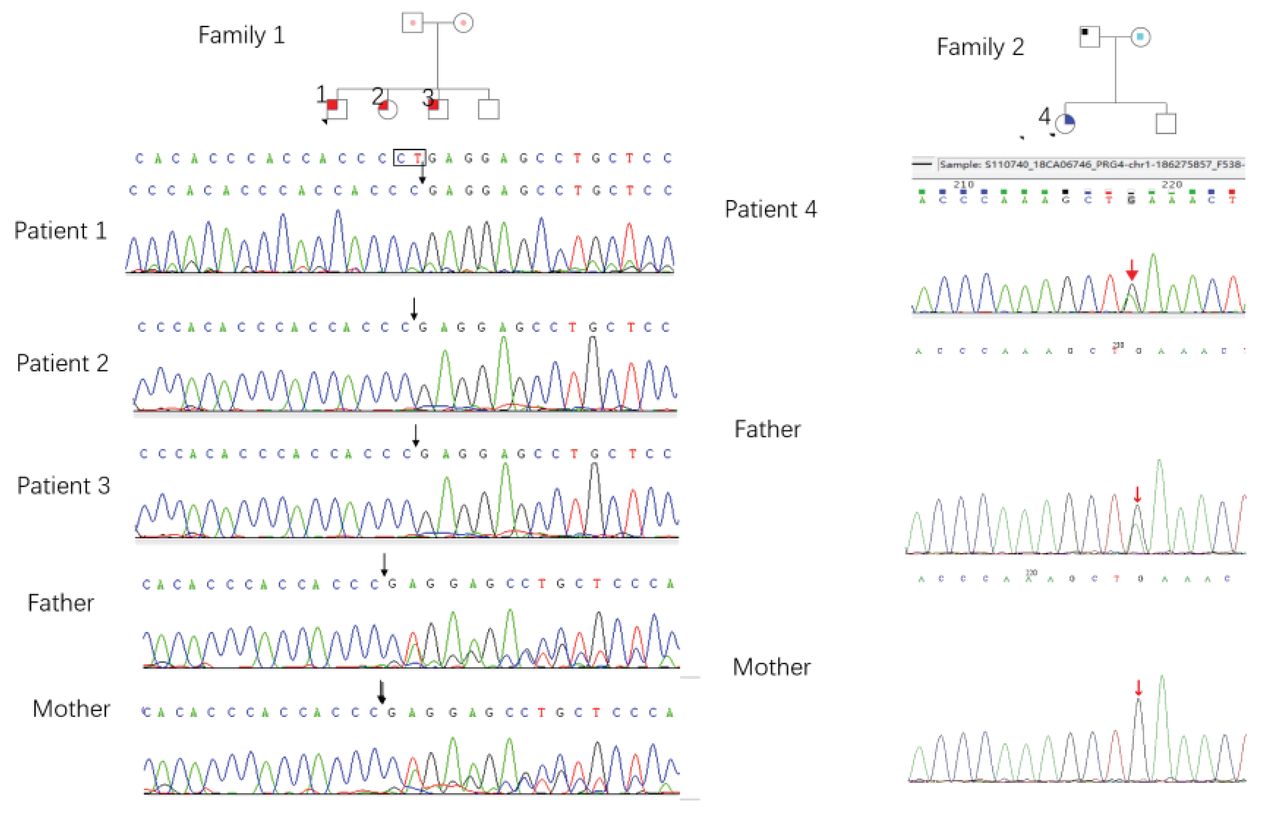 Figure.
Figure. Pedigrees of families 1 and 2 are shown with wild-type or mutant PRG4 alleles. Square, male; circle, female; filled shape, affected subjects; unfilled, unaffected. Arrows indicate the prohead. Dots indicate the gene carriers.
Patient 1 is a 2-month-old male who presented with cystic masses and 20 days of joint swelling. Physical examination indicated swelling in the wrists and knees, and cystic masses in the wrists and ankles; there were no abnormalities in acute-phase reactants or autoantibodies. Plain films indicated roughened bone cortex of the distal radius and slightly reduced bone mineral density of the pelvis. Magnetic resonance imaging (MRI) of the right ankle exhibited nodular signals, suggesting irregular effusion. PRG4 gene sequencing revealed a homozygous missense mutation: c.1982_1983delCT (p.P661Rfs*17). At the 7-month follow-up, the child’s thumbs exhibited camptodactyly.
Patient 2 is the 11-year-old sister of patient 1. She had cystic masses on her wrists and ankles at 39 days old, and swelling in the knees at age 3 years. Physical examination showed camptodactyly, swelling, and cystic masses in the wrists, knees, and ankles. Plain pelvic films indicated shortening of the femoral neck. Hip MRI indicated irregular cystic foci along the muscles of sciatic bodies. Chest computed tomography showed pleural effusion. Right wrist MRI indicated multiple cystic and nodular long T2 signals, suggesting the existence of irregular effusion. A cystic mass biopsy showed chronic granulomatous inflammation. PRG4 sequencing indicated that patient 2 also had the c.1982_1983delCT (p.P661Rfs*17) mutation.
Patient 3 is the 7-year-old brother of patient 1. He developed joint swelling and cystic masses at 5 years of age. He was misdiagnosed as having juvenile idiopathic arthritis and was ineffectively treated with oral methotrexate at another hospital. Physical examination revealed joint swelling and cystic masses in the wrists and ankles. Hip MRI indicated joint effusion and cyst formation, as well as bilateral sciatica with muscle spasms. Plain films showed cervical lordosis. PRG4 sequencing revealed the familial c.1982_1983delCT (p.P661Rfs*17) mutation.
Patient 4 is a 7-year-old female who developed limb ankylosis and intermittent rashes with pruritus after birth. The patient had scattered red rashes on the face, especially around the eyes. No abnormalities in inflammatory indexes and antibodies were observed. Hip MRI showed a short, thick femoral neck, and a large neck trunk angle. Echocardiography showed mild pulmonary stenosis and mitral regurgitation. Skin biopsy revealed no specific manifestations. PRG4 sequencing revealed 2 heterozygous mutations: c.1006G>A (p.E336K) and c.3412a>G (p.m1138v). The patients’ clinical characteristics are summarized in the Table.
Table.Clinical and radiological features at diagnosis of 4 patients from 2 families with CACP.
Molecular defects associated with CACP syndrome occur in the PRG4 gene, which is located on chromosome 1q25-31.4PRG4 encodes lubricin, a protein expressed in joint synovium, cartilage, and nonskeletal tissues (eg, pericardium and pleural cavity).5,6 Neonatal mice lacking lubricin have normal joints, but with age, progressive loss of chondrocytes, hyperplasia of surrounding synovial intimal cells, and degeneration of the osteoarticular joints occur,7 all of which are consistent with CACP. In our study, patient 2 had the most severe clinical presentation, with camptodactyly and simultaneous involvement of the hip and other large joints, whereas patient 1 had only wrist and knee involvement. Seven months later, patient 1 also developed camptodactyly. Yilmaz et al reported that the clinical presentation of CACP worsened over time because of long-term cumulative mechanical stress, according to genotype-phenotype analysis.3,8 New manifestations of CACP syndrome are frequently reported, such as congenital cataracts and protein-loss enteropathy.9,10 Although rashes are unusual presentations, patient 4 had scattered red rashes all over her face; skin biopsy revealed hyperkeratosis, irregular thickening of the acanthocyte layer, perivascular lymphocytic infiltration, and occasional melanin granules and eosinophils. These results suggest that clinical manifestations of CACP are not restricted to the joints, lungs, and heart.
These patients from China with CACP syndrome have previously unreported clinical manifestations (eg, joint cystic masses and rashes) that should be differentiated from Blau syndrome. Patients with Blau syndrome have progressive flexion contracture of the fingers but no hip varus or pericarditis. Further, there are no reports of uveitis in CACP syndrome. Clinicians should not rely solely on genetic testing for CACP syndrome; they also need to carefully assess a patient’s clinical manifestations. We believe that this study presents new thinking on the clinical manifestations of CACP syndrome that may facilitate early diagnosis and encourage follow-up fertility guidance for affected families.
ACKNOWLEDGMENTThe authors thank the patients and their families for participating in this study.
FootnotesJ.M. Zhang and F.Q. Gao contributed equally to this work.
This study was supported by the Special Fund of the Pediatric Medical Coordinated Development Center of Beijing Hospitals Authority (XTCX201819).
The authors declare no conflicts of interest relevant to this article.
Copyright © 2022 by the Journal of Rheumatology
留言 (0)