
記住我
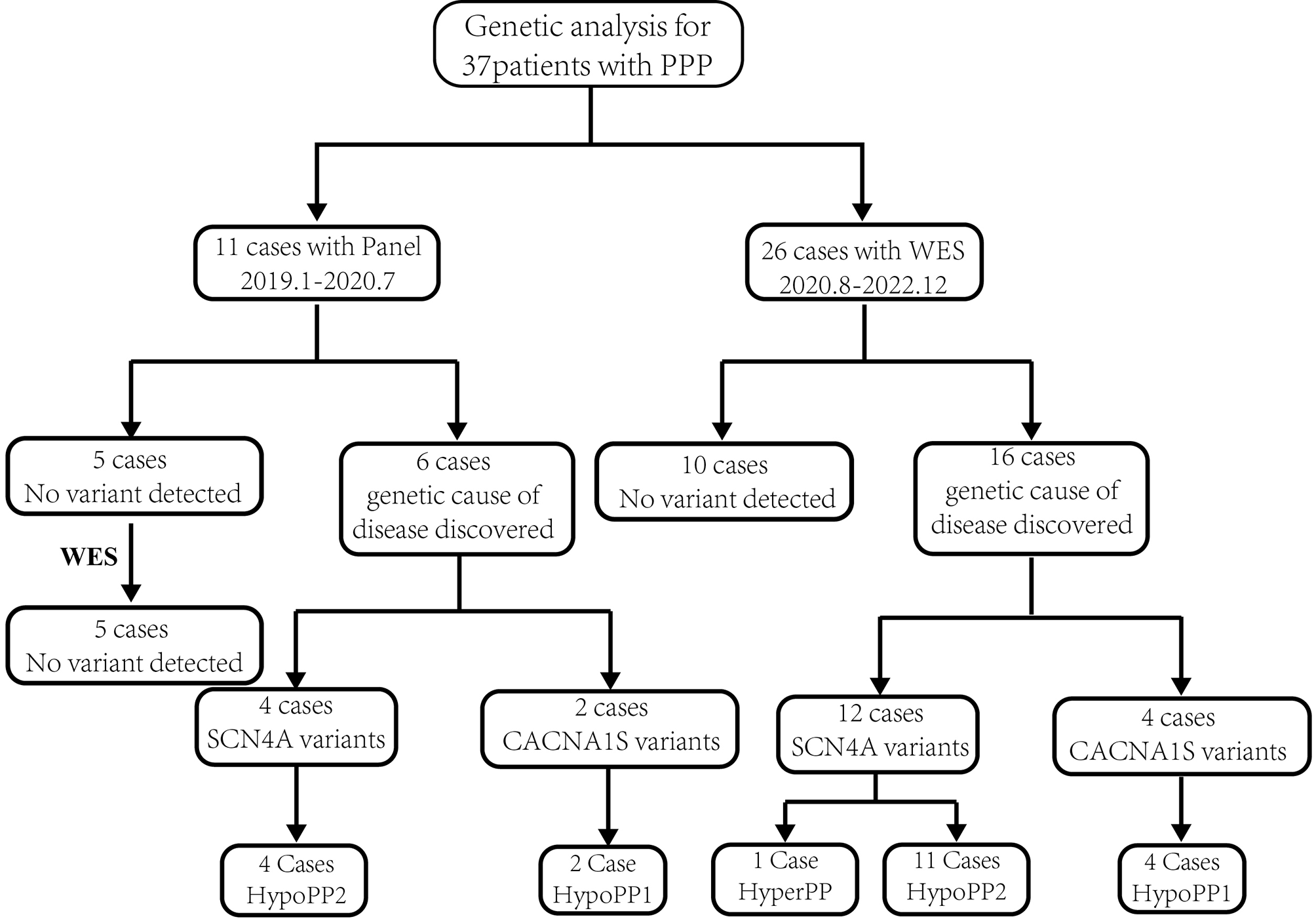


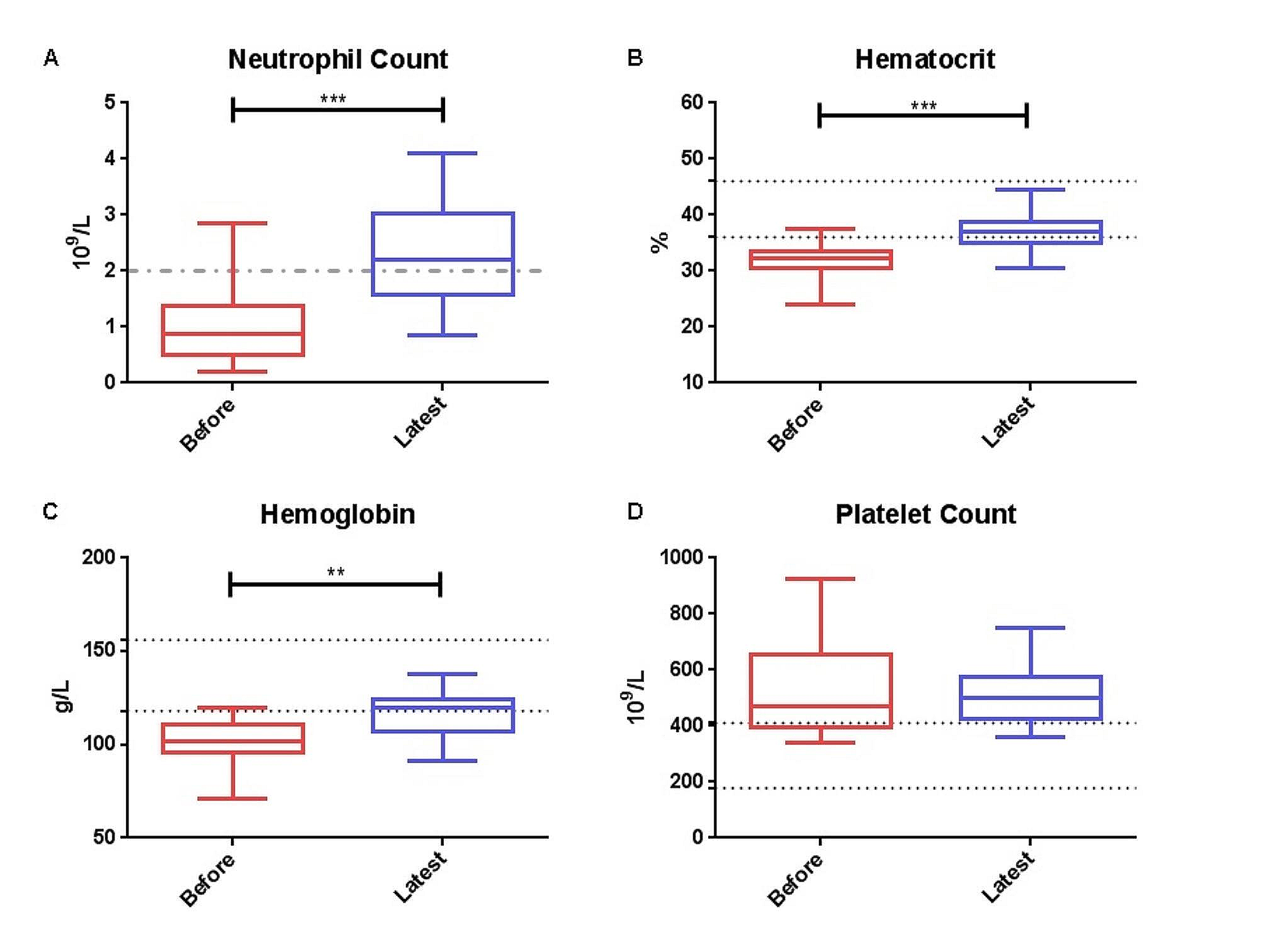

To date, 33 mutations in TAT from 42 families (74 patients) have been described to cause tyrosinemia type II (Table 1, Additional file 1: Fig. 1A) [5,6,7,8,9,10,11,12,13,14,15,16,17]. Missense variants illustrate the predominant variant type (n = 21) in the most recessively inherited autosomal diseases. Furthermore, 5 frameshift variants, 4 nonsense variants, 2 variants that primarily cause splicing site, and 1 skipping complete exon (large deletion) are reported (Fig. 1B). No deep intronic variants and a variant in exon 1 have been reported. The majority of variants are specific. The most frequently observed variant is p.Arg57Ter, which is a nonsense variant causing premature termination of translation, found in 10 patients from 3 families (Table 1). In all patients, this strongly inactivating variant is correlated with high-untreated plasma tyrosine levels. The variant was observed in different ethnicities including Italian, Scottish, and Native American patients. The second most frequently reported variant is p.Thr408Thr, which was found in 7 patients, of 2 unrelated Arab families (Table 1). It is a silent exonic transversion variant, which caused complete missplicing by exon 11 skipping. This skipping leads to the in-frame deletion of 99 nucleotides (33 amino acids). Therefore, the mRNA stability would be affected by this deletion. It is confirmed that this deleterious variant, is responsible for the severe manifestation in these two families. Other frequently seen missense variants are p.Cys151Tyr, p.Leu273Pro, and p.Pro406Leu, which were found in 4 patients with 2 different families, respectively (Table 1). In silico analysis of p.Cys151Tyr variant demonstrated a strong damaging effect corresponding to a deleterious impact on TAT proteins. In addition, p.Leu273Pro variant was reported to affect the stability of TAT protein either by less interaction of protein surface or the reduction of stability of tertiary structure of the protein. The rest of the variants identified in tyrosinemia type II are very rare and reported in one family (Table 1). Variants are found in all exons except exon 1, however, the distribution of missense variants across the TAT gene is not uniform (Fig. 1A). Interestingly, the check-in location of variants revealed that the first variant arises at residue 57 (p.Arg57Ter, Table 1, Fig. 1A). It is demonstrated that probably the amino acids of the N-terminal to this residue are poorly conserved. The most common accumulation of variants in this gene occurs in exon 11, which might be particularly due to the susceptibility of this region to mutation. See additional file 1 for the more information in reported patients with tyrosinemia type II and III.
Table 1 Summary of reported variants in TAT gene of patients with tyrosinemia type IIFig. 1
Schematic illustration (not to scale) of the TAT gene and location of sequence variants associated with Tyrosinemia type II deficiency. A Exon (boxes)–intron (lines) structure of the human TAT gene. Coloring boxes are consistent with panel. B Pie chart summarizing types of TAT variants reported to date
Regarding the geographical distribution of variants, the majority of reported variants are patients from Europe, North America, Japan, Tunisia, and Palestinian Arabs. For the most prevalent variant in TAT gene, p.Arg57Ter, the founder effect is apparent in northern Italy, which should be analyzed in other populations of Mediterranean ancestry. Little information about the epidemiology and molecular defects in tyrosinemia type II patients from East Asia is available at this time. To the best of our knowledge, no mutation in the TAT gene has been reported from Central America, Africa, the Middle East, and the Oceania continent.
4-Hydroxyphenylpyruvate dioxygenase (HPPD) geneEleven disease-causing variants in 16 patients are recently reported in HPPD gene [4, 18,19,20,21,22,23]. All 11 HPPD alleles are divided into 7 missense variants, 2 nonsense variants, 1 splice defect, and 1 frameshift variant (Table 2). Nine of them are in the exonic region and 2 out of 11 variants are intronic ones. All variants are unique, except p.Tyr160Cys, which is a missense variant found in 2 different patients from 2 families (Table 2). The analysis of the crystal structure of Pseudomonas enzyme demonstrated that the Tyr160 residue corresponds to a position in an alpha helix, which is involved in inter-subunit contacts. Therefore, the position of variant might be important for the stability of the enzyme, but this needs to be further elucidated. Three variants were reported in exon 11 and 2 variants in the intronic region of 10, and 11. Therefore, the largest accumulation of variants happens in this region, which is suggestive of its susceptibility to mutation.
Table 2 Summary of reported variants in HPPD gene of patients with tyrosinemia type IIIRegarding the geographical distribution of variants, the majority of reported variants are patients from Europe, and Asia (Japan, China, and Iran). No information about the epidemiology and molecular defects in tyrosinemia type III patients from North and Central America, Africa, Australia, and the Oceania continent are available.
Genotype–phenotype correlationThe correlation between genotype and phenotype is described as the possibility of an association between a specific mutation and/or class of mutation with a special clinical abnormality. Therefore, finding genotype–phenotype correlations in inherited metabolic disorders as rare diseases are difficult and complicated by several factors, such as the absence of large cohorts of patients for analysis, the large phenotypic heterogeneity associated with the same mutation, and a high proportion of private mutations [24]. The great number of variants in TAT and HPPD genes are private variants, so, genotype–phenotype correlations or correlations between type or location of the mutation and clinical manifestation have not been established so far. Therefore, for this purpose, we present a comprehensive list of patients identified from the reported literature, along with clinical and biochemical data (Additional file 1: Table S1). This helped us not only to precisely estimate the number of published patients with tyrosinemia type II and III diseases but also helped to stratify patients for genotype–phenotype correlations.
Data on clinical and biochemical results of tyrosinemia type II were available for 70 patients. We have identified among them, only 7 patients had elevated levels of tyrosine along with detected pathogenic variants, however, clinical manifestations were asymptomatic (Additional file 1: Table S1). Approximately 90% of patients had correlated clinical and biochemical manifestations with the type of detected variant.
Nevertheless, 5 out of 16 patients from 4 different families and populations with tyrosinemia type III were asymptomatic even though they had elevated tyrosine levels and high urinary excretion of 4-HPL and 4-HPP. However, due to the small number of patients, it is not possible to conclude clearly. It needs further investigation.
留言 (0)