
記住我





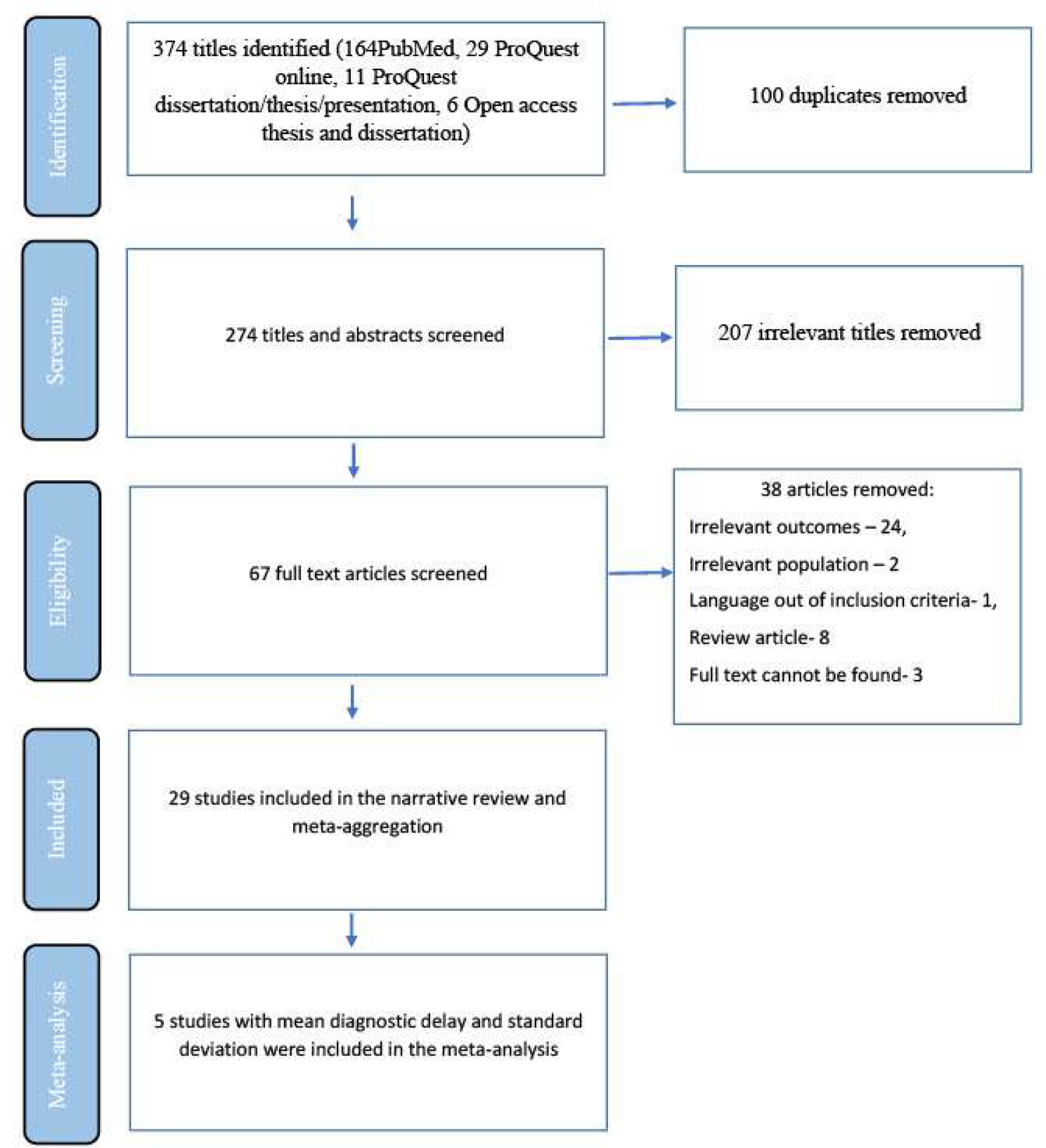
To begin determining ENPP1 Deficiency prevalence estimates, ENPP1 variants were identified through a comprehensive literature review performed using the Mastermind Genomic Search Engine [8]. A total of 183 ENPP1 variants were identified and interpreted according to the American College of Medical Genetics and Association of Molecular Pathologists (ACMG/AMP) variant interpretation guidelines [9]. Of these variants, 77 were classified as Pathogenic/Likely Pathogenic, 91 as Variants of Undetermined Significance (VUS), 13 as Benign/Likely Benign and 2 as conflicting (having sufficient evidence to meet both a Benign and Pathogenic classification). All Benign/Likely Benign and conflicting variants were excluded from the proceeding analysis.
Variants classified as Pathogenic/Likely Pathogenic or VUS that had a non-zero allele frequency in the Genome Aggregation Database (gnomAD) were included or excluded as described in Additional file 1: Figs. S1 and S2, respectively [10]. This selection process considered the variant’s presence in ENPP1 Deficiency patients, allele frequency and presence in homozygotes in gnomAD, classification in ClinVar, effect type, and predictions from computational algorithms [11]. Following this process, a total of 27 Pathogenic/Likely Pathogenic variants and 17 VUSs were included in the prevalence calculation as Known Pathogenic/Likely Pathogenic variants and VUS variants, respectively (Fig. 1; Additional file 2: Table S1). Notably, 49 of the total Pathogenic/Likely Pathogenic variants (64%) and 69 of the total VUS (76%) were excluded solely as a result of not being present in gnomAD.
Fig. 1
Inclusion/Exclusion of Variants in ENPP1. The number of variants included or excluded, along with the reason, is displayed. Loss of function (LOF) variants include start loss, nonsense, frameshift, and canonical splice site variants. VUS Variant of Undetermined Significance
In addition, there were variants in the gnomAD database that were not identified in the published literature, which were included or excluded as described in Additional file 1: Fig. S3. This selection process considered the variant’s allele frequency and presence in homozygotes in gnomAD, classification in ClinVar, effect type, and predictions from computational algorithms. Following this process, a total of 42 Presumed Pathogenic Loss of Function (LOF) variants and 128 Presumed Pathogenic missense variants were included in the prevalence calculation (Fig. 1; Additional file 2: Table S1).
The overall allele frequency in gnomAD was summed across the specific groups of included variants—Known Pathogenic/Likely Pathogenic, VUS, Presumed Pathogenic LOF, and Presumed Pathogenic Missense variants. The carrier frequency and genetic prevalence was then calculated using the Hardy–Weinberg equilibrium equation for these groups to showcase the range of estimates that result from different levels of stringency in the variant selection process.
The carrier frequency for ENPP1 variants associated with ENPP1 Deficiency was estimated to be 1 in 509 to 1 in 127 in the general population which corresponds to a genetic prevalence of 1 in 1,033,927 to 1 in 64,035 pregnancies (Table 1).
Table 1 Genetic Prevalence of ENPP1 DeficiencyThere was a 1,515% difference between the lowest, most conservative prevalence estimate, which included only variants known to be Pathogenic/Likely Pathogenic by ACMG/AMP, and the highest, most inclusive prevalence estimate, which included all Presumed Pathogenic variants as well as VUS. Notably, the inclusion of Presumed Pathogenic LOF and Presumed Pathogenic Missense variants had a large impact on the estimated prevalence. Inclusion of Presumed Pathogenic LOF variants increased the estimate (excluding VUSs) by 209% which increased by another 304% with inclusion of Presumed Pathogenic missense variants. In contrast, inclusion of VUS had a comparatively minor impact on the estimated prevalence—a 29–119% increase.
Our estimated genetic prevalence was also 211% higher than a previous study which estimated it to be ~ 1 in 200,000 pregnancies, including all presumed Pathogenic variants [3]. This study had several methodological differences including the use of a smaller population database (the Exome Aggregation Consortium (ExAC)), not using Mastermind to complete a literature review, and no use of ACMG/AMP interpretation [12]. Because of the greater number of individuals represented in gnomAD as compared to ExAC, the number of carriers identified was likely increased based on a more representative statistical sampling for these rare variants. Moreover, a significant number of new variants and patients were reported since the prior publication, resulting in both a greater number of Known Pathogenic/Likely Pathogenic variants (27 compared to 17) as well as an increased number of Presumed Pathogenic variants (170 compared to 96).
Specifically pertaining to the Known Pathogenic/Likely Pathogenic variants, there were 8 variants that were presumed to be Pathogenic in the previous study by assessment of computational predictions but confirmed to be Pathogenic/Likely Pathogenic by ACMG/AMP standards in this study, 3 variants that were classified as Pathogenic/Likely Pathogenic in both studies but were present only in gnomAD, 3 variants that were classified as Pathogenic in the previous study but were VUS by ACMG/AMP in this study, 2 that were presumed to be Benign in the previous study but were found to be Likely Pathogenic in this study, and 1 variant that was not identified in the previous study but was found to be Pathogenic/Likely Pathogenic in this study.
Overall, the assessment of complete literature evidence along with ACMG/AMP interpretation was instrumental in ensuring the precision of the genetic prevalence estimate. If the estimate relied solely on classifications in ClinVar, but otherwise used the same methodology, a total of 15 variants would have been excluded on the basis of conflicting classifications and/or conflicting computational predictions.
Assessing the contribution of individual ENPP1 variants to the genetic prevalence estimate revealed that only 13 variants accounted for 50% of the total allele frequency included in the prevalence calculation (Fig. 2). The top five most frequent variants alone—c.26dup; p.Gly10ArgfsTer67 (0.032% allele frequency), c.2114C > T; p.Thr705Met (0.025% allele frequency), c.2236A > C; p.Asn746His (0.021% allele frequency), c.2713_2717del; p.Lys905AlafsTer16 (0.021% allele frequency), c.1352A > G; p.Tyr451Cys (0.016% allele frequency)—accounted for 30% of the total.
Fig. 2
Contribution of Individual ENPP1 Variants to the Genetic Prevalence Estimate
The most common variant (c.26dup; p.Gly10ArgfsTer67) is a previously unpublished variant that is presumed to be pathogenic as a result of it being a frameshift variant at the 5’ end of the gene but may have an inflated allele frequency due to the small number of captured alleles (3,086 overall). The remaining high frequency variants were unaffected by this issue. p.Thr705Met is an unpublished variant that is predicted to be pathogenic by computational algorithms, p.Asn746His has previously been found in four probands initially suspected of having X-Linked Hypophosphatemia (XLH) but is considered a VUS by both our interpretation and interpretations in ClinVar, and p.Lys905AlafsTer16 and p.Tyr451Cys have been found in multiple patients with GACI/ARHR2 and are interpreted as Pathogenic and Likely Pathogenic, respectively [13,14,15,16].
In addition, the estimated heterozygous carrier frequency of ENPP1 variants was found to vary between specific populations in gnomAD, with the East Asian population having a significantly higher carrier frequency than other populations (2.3%; Fig. 3). The most frequent variant in the East Asian population is c.26dup; p.Gly10Argfs*67 (0.32% allele frequency in the East Asian population). As noted previously, this variant was captured in a low number of alleles overall, and 1/310 alleles in this population, which could have artificially inflated the allele frequency, and subsequently the carrier frequency. Removing this variant from consideration, the carrier frequency of ENPP1 variants in the East Asian population is 1.6%, which remains higher than other populations in gnomAD.
Fig. 3
Population-Specific Carrier Frequencies of ENPP1 Variants. The carrier frequency was calculated using the Hardy–Weinberg equilibrium equation and the sum of the allele frequencies of the specified variants. Known Pathogenic/Likely Pathogenic: variants that were classified as Pathogenic/Likely Pathogenic by ACMG/AMP. Presumed Pathogenic LOF: start loss, nonsense, frameshift, and canonical splice site variants that were found in gnomAD but not the published literature. Presumed Pathogenic Missense: predicted damaging missense variants that were found in gnomAD but not the published literature
Notably, considering only the Known Pathogenic/Likely Pathogenic variants, the carrier frequency of ENPP1 variants was highest in the Finnish population at 0.43%, which the next highest being the East Asian population at 0.32% (Fig. 3). The most frequent pathogenic variant in the Finnish population is c.2713_2717del; p.Lys905Alafs*16 (0.18% allele frequency in the Finnish population), which has previously been published in three Caucasian siblings with GACI as well as three unrelated patients with GACI/ARHR2, two of which were American (unspecified ethnicity), and one of which was Finnish [3, 14, 16].
留言 (0)