
記住我
In situ vaccination with cowpea mosaic virus (CPMV) has potent antitumor efficacy at local treated tumors.
More insight is needed to delineate potency and mechanism CPMV-in situ vaccination (ISV) (as solo or in combination with checkpoint therapy) of the systemic antitumor immunity and abscopal effect.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY:By activating DCs and T cells with agonistic CD40 antibody and programmed death 1 antibody, we developed a potent ISV combination with CPMV for uncommon exceptionally immune cold mouse tumors that minimally respond to CPMV only.
The overall ISV strategy and specifically the use of CPMV for ISV provides a powerful platform for local and systemic solid tumor immunotherapy.
BackgroundIn situ vaccination (ISV) is increasingly recognized as a valuable treatment strategy in cancer immunotherapy. The appeal of cancer vaccination revolves around vaccines generating tumor antigen specific immune responses. Standard vaccines combine and deliver antigens for recognition by lymphocytes along with immune adjuvants to stimulate innate immune cells to focus immune recognition and response on the vaccine antigens. ISV circumvents the need to identify tumor antigens by local treatment of an identified tumor with immune adjuvant, cytokines, immune manipulating antibodies or cytotoxic immunostimulatory manipulations like radiation or heat. The immune stimulation in the treated tumor reverses local tumor-mediated immunosuppression and produces an immunostimulatory tumor microenvironment (TME). When optimal, this leads to successful attack of the tumor by various leukocytes resulting in the release of both neoantigens and tumor-associated antigens, effective presentation of those antigens by antigen-presenting cells (APCs), and generation of a greatly expanded systemic adaptive immune response that is patient-specific and able to control tumors that were not directly treated with ISV.1 2 In ISV, the antigen is in the cancer cells themselves and the treatment essentially delivers adjuvant to reverse the tumor-mediated immunosuppression that blocks antitumor immunity.
We have been investigating cowpea mosaic virus (CPMV) as an ISV antitumor agent. CPMV, a plant virus that does not infect animal cells, is extremely immunostimulatory; its assembled capsids are recognized by toll-like receptors (TLRs) 2 and 4, its single strand RNAs (ssRNAs) stimulate TLR7 .3 4 Depending on the model studied, the treatment relies on various combinations of neutrophils, APCs, adaptive immune cells, interleukin (IL)-12, type I interferons and/or interferon (IFN)-γ.5 6 ISV with CPMV alone remodels the TME and stimulates a potent antitumor immune response in mouse models of melanoma, ovarian cancer, breast cancer, colon cancer, and glioma6–8 as well in companion dogs with multiple spontaneous cancers.9
ISV is a local treatment that involves direct treatment of an identified tumor but is not fundamentally a local therapy. Optimal ISV stimulates systemic antitumor immunity that directly impacts untreated metastatic disease, with or without other immunotherapies. Although CPMV elicits strong local antitumor efficacy that slows tumor growth, with many models and depending on tumor size and number of treatments delivered, the tumor is often temporarily suppressed but not eliminated.2 10 Recently, the combination of inactivated CPMV and OX40 agonist exhibited potent antitumor efficacy in the bilateral melanoma mouse model,11 however, the systemic efficacy of CPMV in suppressing tumors not directly treated with ISV has not been thoroughly investigated.
Advances in immune checkpoint blockade (ICB) therapies, including against programmed death 1 (PD-1) or its ligand (PD-L1) has established immunotherapy as a viable treatment of multiple solid different tumor types.12 However, many patients do not respond at all or develop resistance to ICB with anti-PD-1 (aPD-1)/anti-PD-L1 (aPD-L1) after initial tumor regression,13 which is often attributed to poor T-cell infiltration into the tumors.14 There is an urgent need for the development of novel approaches that work with checkpoint blockade to increase the fraction of patients that achieve long-term remission.
A recent study highlights the synergy of CPMV ISV and checkpoint blockade. Intratumoral CPMV treatment sensitizes TME to immune checkpoint therapy, that is, by upregulation of PD-1/PD-L1 and OX40 expression and by remodeling the TME. Combination therapy of CPMV and OX40 agonist or aPD-1 showed dramatic increases in efficacy in multiple tumor models. Depletion of either CD8+ or CD4+ T cells significantly abrogated the efficacy demonstrating that CPMV ISV elicits adaptive antitumor immunity.10 We built on this work and study the mechanism and temporal changes in immune cells and cyto/chemokines—particularly, we focus on systemic antitumor immunity and abscopal effect using two-tumor mouse models.
Of particular interest was also to investigate the role of dendritic cells and add an agonist that activates CD40 on dendritic cells (DCs) among other APCs. Recent evidence indicates there is a critical role for tumor-residing Batf3-dependent conventional type-1 dendritic cells (cDC1s) in priming and expanding tumor-specific CD8+ T cells and their recruitment to the TME.15–19 cDC1 cells efficiently cross-present tumor antigens.20 Studies reveal that activation of cDC1s improves overall responses to ICB therapies with aPD-1/PD-L1.21 22
Activation of CD40 provides potent maturation and anti-apoptotic signals to DCs and other APCs, which enables more effective activation of cytotoxic T lymphocytes, induces IL-12 production, and overcomes T-cell tolerance.23 24 Moreover, combination of TLR agonist and CD40 agonist synergizes to enhance CD8+ T-cell expansion,25 and mediate potent antitumor immunity in multiple syngeneic mouse models.26–28 In recent reports, agonistic anti-CD40 has been applied in situ and illustrated greater antitumor efficacy with radiation or focused ultrasound heating.29 30 Since CPMV strongly induces IL-12, is a multi-TLR agonist,3 6 there is rationale for combining CPMV and checkpoint blockade with CD40 stimulation as a new ISV regimen to achieve better outcomes.
In our current study, we investigate whether CPMV ISV generates systemic antigen-specific antitumor immunity that can impact untreated tumors (abscopal effect), and whether ISV with CPMV combined with agonistic anti-CD40 (aCD40) facilitates the priming, expansion, and infiltration of tumor-specific CD8+ T cells into cold TME and increases response to aPD-1 therapy. The studies reveal that, in multiple mouse models of poorly T cell-infiltrated and aPD-1-resistant tumors, CPMV induces significant systemic immune-mediated efficacy by itself. CPMV ISV outcomes are further improved when combined with both local agonistic CD40 antibody and systemic ICB therapy. This combination caused consistent complete regression not only of ISV treated but also untreated distant tumors.
MethodMiceFemale C57BL/6 mice and Batf3−/− mice on C57BL/6 mice background were purchased from the Jackson Laboratories and bred in house, and Rag2 knockout mice were gifts of Prof Yina Huang of Dartmouth Geisel School of Medicine. Female BALB/c−AnNCr mice were purchased from Charles River Laboratories. All mice were age matched (7–12 weeks old) at the beginning of each experiment and kept under specific pathogen-free conditions and housed in the Laboratory Animal Resources. All animal studies were approved by the Institutional Animal Care and Use Committee of Dartmouth College under approved protocol 2137.
Cell linesThe 4T1 breast cancer, MC38 colon adenocarcinoma, CT26 colorectal carcinoma, and B16-F10 (B16) melanoma cell lines were purchased from the American Type Culture Collection. 4T1, MC38, and B16 cells were cultured in RPMI (Gibco) supplemented with 10% fetal bovine serum, 1% nonessential amino acids, 2 mM l-glutamine, 0.5% penicillin/streptomycin, and 55 µM 2-mercaptoethanol. These cell lines were authenticated by morphology, phenotype, and growth, and routinely screened for Mycoplasma, and were maintained at 37°C in a humidified 5% CO2 atmosphere.
Production of CPMVCPMV was propagated in Vigna unguiculata (black-eyed pea No. 5) plants. Primary leaves were mechanically inoculated with 0.1 mg/mL CPMV in phosphate-buffered saline (PBS). Virus particles were purified using chloroform: butanol extraction and polyethylene glycol precipitation combined with sucrose gradient ultra-centrifugation. Detailed protocols for propagation and purification of CPMV have been described previously.31 Purified CPMV was characterized using gel electrophoresis, size-exclusion chromatography, transmission electron microscopy and ultraviolot-visible light spectroscopy (online supplemental file 1).
Tumor inoculationB16F10 (1.25×105) or 4T1 (1.25×105), or MC38 (1.25×105) tumor cells were injected in 30 µL PBS intradermally on both flanks under anesthesia with isoflurane. We use intradermal tumor growth so we can directly visualize the tumors and enable accurate and complete intratumoral injection of reagents. For two-tumor mouse models, the treated tumor was inoculated on day −7, the untreated tumor on day −4 and the treatment was started on day 0. Growth curves stop whenever the first mouse in the group reaches the end point (1000 mm3), since that immediately changes the growth curve average on a given day, but the outcome is completely tracked by survival curves.
ISVTumor-bearing mice were treated with CPMV (100 µg/dose) and/or aCD40 Ab (10–20 µg/dose) in 30 µL PBS or control PBS injected intratumorally as scheduled in Figures.
Tumor growth was measured every other day, and the volumes were calculated by determining the length of short (w) and long (L) diameters (volume =w2×L/2). Experimental endpoints were reached when tumors exceeded 15 mm in diameter or when mice displayed symptoms of poor health.
In vivo antibody treatmentFor PD-1 blockade, aPD-1 Ab (Bio X Cell), or rat IgG2b (clone LTF-2, Bio X Cell) were given intraperitoneally (i.p.) every third day from the first day ISV was performed at a dose of 200 µg/mouse. For in vivo depletion of lymphocytes, 200 µg of anti-CD4 (clone GK1.5, Bio X Cell), anti-CD8β (clone Lyt 3.2, Bio X Cell), or rat IgG2b (clone LTF-2, Bio X Cell) were injected i.p. every third day three times from the first treatment day. For in vivo depletion of IL-12 and IFN-γ, 1 mg of anti-IL-12p40 (clone C17.8, Bio X Cell) and anti-IFN-γ (clone R4−6A2, Bio X Cell) Abs were administrated by i.p. injection at the first treatment day, with follow-up doses of 500 µg for five consecutive days.
Flow cytometrySingle-cell suspensions of mouse lymph nodes were prepared for flow cytometric analysis. Fc receptors were blocked with anti-mouse CD16/32 (BioLegend) and surface stained with indicated markers. We used the published gating strategy to dissect the myeloid and lymphoid compartment changes with Bio-Rad ZE5 Cell Analyzer and FlowJo V.10.8.1.15
Statistical analysisAll results are expressed as means±SEM (n=3–5) as indicated. Student’s t-test was used to compare the statistical difference between two groups, and one-way or two-way analysis of variance with Sidak’s or Tukey’s multiple comparison tests were used to compare three or more groups (*p<0.05, **p<0.01, ***p<0.0005, ****p<0.0001). Survival rates were analyzed using the log-rank (Mantel-Cox) test (**p<0.01). GraphPad Prism V.8.0.2 software was used to calculate significance between the samples. FlowJo V.10.8.1 was used to analyze corresponding data. All experiments were repeated, and representative figures were presented unless otherwise noted. P values≤0.05 were considered significant. Statistical test is indicated in each figure.
ResultsISV of CPMV generate systemic efficacy in various tumor modelsBoth local and abscopal efficacy of CPMV were assessed in four different syngeneic mouse tumor models: B16F10 melanoma, 4T1 triple negative breast cancer, MC38 colon adenocarcinoma, and CT26 colorectal carcinoma. The treated tumor was inoculated on day −7, the untreated tumor was inoculated on day −4 and treatment was started on day 0 (figure 1A). CPMV had significant efficacy on both treated and untreated tumors in B16F10, MC38 and CT26 models (figure 1B–D); however, CPMV did not elicit local or abscopal tumor inhibition in 4T1 (figure 1E). This 4T1 treatment resistance correlates with the finding that 4T1 recruits exceptional numbers of myeloid-derived suppressor cells and is quite resistant to immunotherapy.32 33
 Figure 1
Figure 1 In situ vaccination with CPMV generates systemic antitumor immunity in B16F10, MC38, CT26, but not in 4T1 (A) Schematic presentation of experimental setup. Two-tumor bearing mice of B16F10, MC38, CT26 had first tumor intradermal inoculated at day −7 and the distant tumor inoculated on day −4, then day −7 tumor treated with 2 weekly intratumoral injections of CPMV. (B–E) Growth curves for corresponding tumors. Two growth curves were analyzed using two-way analysis of variance, with p>0.05 as ns, p<0.05 as *, p<0.01 as **, and p<0.001 as ***. All experiments were repeated at least once with similar results and each with n=3–5 mice/group. CPMV, cowpea mosaic virus; I.D., intradermal; PBS, phosphate-buffered saline.
Systemic efficacy by CPMV requires CD8+ T cells and is antigen specificWe investigated the cellular mechanisms that account for systemic efficacy of ISV using CPMV. CPMV-elicited immune changes were evaluated with flow cytometry using tumor draining lymph nodes (TdLN). CPMV-treated mice had a significantly increased fraction of CD8+ T cells in TdLN (figure 2A) and this agrees with our previous findings of CD8+ T-cell increases in the treated solid tumors as well as in the peritoneum in a dispersed peritoneal ovarian tumor model.5 10 34 Thus, we hypothesized that CD8+ T cells are required for the systemic efficacy elicited by CPMV.
 Figure 2
Figure 2 Systemic efficacy by CPMV requires CD8+ T cells and is antigen specific. (A) Tumor draining lymph nodes from both CPMV-treated and PBS-treated mice were collected 48 hours after first treatment injection. Frequencies of CD8+ T cells among CD45+ cells in B16F10-bearing mice in tumor draining lymph nodes. (B) Tumor growth curves for CPMV-treated and PBS-treated B16F10 in Rag2−/− mice. (C) CD4− or CD8− depleted treatment of B16F10 in normal C57BL/6J. (D) Unmatched two-tumor-bearing mice with B16F10 (treated) and MC38 (distant) tumors in normal C57BL/6J. Data for bar graphs calculated using unpaired Student’s t-test. Flank tumor growth curves were analyzed using two-way analysis of variance, with p>0.05 as ns, p<0.05 as *, p<0.01 as **, and p<0.001 as ***. All experiments are repeated at least once and each with n≥3 mice/group. CPMV, cowpea mosaic virus; PBS, phosphate-buffered saline.
To test the requirement for lymphocytes, we implanted the tumors in Rag2−/− mice, which lack B and T cells, and we found the abscopal efficacy by CPMV was completely abolished in the untreated tumor, despite normal response of the treated tumor (figure 2B). To more precisely identify cell types involved in CPMV local and systemic ISV response, we used depleting antibodies against CD4 and CD8 to further define the contributing T-cell populations. CPMV treatment had reduced local efficacy and completely lost systemic efficacy in CD8-depleted animals. CD4-depletions did not affect either local or abscopal responses (figure 2C). This supports CD8+ cells as necessary for CPMV-elicited systemic efficacy, while CD4 cells may not be required.
Systemic efficacy of CPMV is antigen specificSince the systemic efficacy of CPMV requires CD8+ cells, we next focused on whether this efficacy is antigen specific. Since CPMV elicits abscopal effects in many different models (figure 1), we tested antigen specificity of the response using unmatched syngeneic tumors at each flank (figure 2D). The syngeneic tumor cell line MC38 were used as the untreated tumors and were matched with treated B16F10 tumors. Consistent with tumor-specific T cell-mediated killing, when different syngeneic tumors were used there was no inhibition of the untreated tumor growth, demonstrating that the systemic CD8+ T-cell mediated effects were specifically against antigens in the treated tumor.
Cross presentation by cDCs is required for CPMV-elicited systemic efficacy which is also IFN-γ and IL-12 dependentIn prior studies, we have shown that CPMV is predominantly ingested by phagocytes, including macrophages, DCs, and neutrophils.34 Since antigen specificity is part of the systemic efficacy and phagocytes directly ingest CPMV we examined the effect of CPMV treatment on phagocytic APCs to identify a cell population that may be cross-presenting tumor antigens to the CD8+ T cells controlling tumor growth. We used the published gating strategy to dissect the myeloid and lymphoid compartment changes.15 We found that the cDC1 (CD45+, Ly6C–, MHChi, F4/80–, CD103+) population, an important category of APCs within the TME,35 roughly doubled within 48 hours in TdLN after the second weekly dosing (figure 3A); meanwhile, the composition of cDC2 (CD45+, Ly6C–, MHChi, F4/80–, CD11b+) decreases. This corresponds to our previous findings that proinflammatory cytokines were induced in treated TdLNs.36 cDC1 efficiently cross-present antigens to CD8+ T cells and require the transcription factor BATF3 for development.19 37 Thus, we tested whether cDC1s were necessary for treatment efficacy. Treated tumors in Batf3–/– mice that lack cDC1 responded to CPMV ISV, however response of distant tumors was eliminated (figure 3B), confirming the requirement of cDC1 cells for systemic treatment efficacy. While the experiment was not set up to test whether response to treatment by CPMV with depletion of CD8 (figure 2C) and the same treatment in mice lacking cDC1 were identical, it does appear that both manipulations did reduce the efficacy of response in the treated tumor. This suggests that CD8 T cells and presentation of antigen by cDC1 do play at least a minor role in response of a treated tumor to CPMV.
 Figure 3
Figure 3 Cross presentation by cDC1s is required for CPMV-elicited systemic efficacy (A) Frequencies of DCs, CD11b+DCs, and CD103+DCs among CD45+ cells in B16F10-bearing mice were compared in tumor-draining lymph nodes 48 hours after the second weekly treatment. (B–D). Flank tumor growth curves of CPMV-treated and PBS-treated B16F10 tumors in (B). Batf3−/−, (C). Immune system manipulation using anti-interferon-γ or anti-interleukin-12 depletion antibody correspondingly. Data for bar graphs calculated using unpaired Student’s t-test. Flank tumor growth curves were analyzed using two-way analysis of variance, with p>0.05 as ns, p<0.05 as *, p<0.01 as **, and p<0.001 as ***. All experiments are repeated at least once and each with n≥3 mice/group. aIFNg, anti-interferon gamma; aIL-12, anti-interleukin 12; cDCs, cDCs, conventional dendritic cells; CPMV, cowpea mosaic virus; PBS, phosphate-buffered saline.
A past study has shown that successful aPD-1 treatment requires T cell-DC crosstalk involving IFN-γ and IL-12.38 It is of interest to investigate whether IFN-γ and IL-12 are needed for this ISV using CPMV. Using depleting antibodies for IFN-γ and IL-12, we found that loss of either IFN-γ or IL-12 attenuated the CPMV response in treated tumors and its systemic efficacy is lost (figure 3C). This shows that CPMV-elicited systemic efficacy is IFN-γ and IL-12 dependent.
ISV of CPMV using agonistic aCD40 antibody further facilitates priming, expansion, and infiltration of CD8+ T cells in tumorsSince our data show that DCs play an important role in CPMV-elicited efficacy, we performed studies to determine whether local stimulation of DCs with a CD40 agonist in addition to CPMV ISV would generate a stronger ISV combination. Given the potent role that CD40 can play in APC activation39 and its ability to sensitize DCs to TLR stimulation,40 the effect of an agonistic CD40 mAb on treatment efficacy was investigated in vivo when administered intratumorally along with CPMV (figure 4A). Agonistic CD40 alone did not affect the larger treated tumor but did slow the growth of the smaller untreated tumor. The ISV combination of CPMV and aCD40 generated better efficacy in both treated and distant tumors (figure 4B). On day 12, both treated and distant tumors in dual-treated group were ~40% size of the corresponding tumors in CPMV-treated group; however, the ISV combination using the parameters applied did not eradicate most tumors.3
 Figure 4
Figure 4 In situ vaccination of CPMV with anti-CD40 generates better efficacy. (A) Schematic presentation of experimental setup. Two-tumor bearing mice of B16F10 or 4T1 had their first tumor treated with weekly intratumoral injection of CPMV and/or CD40 agonist antibody (aCD40). (B, C) Growth curves for corresponding tumors. Tumor growth curves were analyzed using two-way analysis of variance, with p>0.05 as ns, p<0.05 as *, p<0.01 as **, and p<0.001 as ***. All experiments are repeated at least once and each with n≥3 mice/group. aCD40, agonist anti-CD40; CPMV, cowpea mosaic virus; I.D., intradermal; PBS, phosphate buffered saline.
Of note, this new ISV combination of CPMV and aCD40 Ab was able to elicit both local and systemic efficacy in 4T1 tumor model (figure 4C). This raised the possibility that adding another treatment would further enhance the antitumor efficacy and hopefully achieve the goal of tumor elimination.
ISV combinations synergize with aPD-1 and elicit potent antitumor immunityResponse to systemic blockade of PD-1/PD-L1 has been associated with the presence of CD8 tumor infiltrating lymphocytes (TILs).14 A previous study in an ovarian cancer model (ID8/Vegf/Defb29) showed the frequency of PD-1-expressing CD8+ TILs increased in tumors and PD-1 expression was more prevalent in treated CD8+ T cells,10 suggesting a possible higher susceptibility to PD-1-mediated inhibition of the ISV-induced tumor-specific CD8+ T-cell infiltrates. This provides the rationale to include aPD-1 as a more powerful combination. Indeed, this new combination of ISV with CPMV/CD40 stimulation along with systemic injection of aPD-1 provided clearly improved systemic efficacy in B16F10 as well as in 4T1 as shown by response of untreated tumors (figure 5A–C).
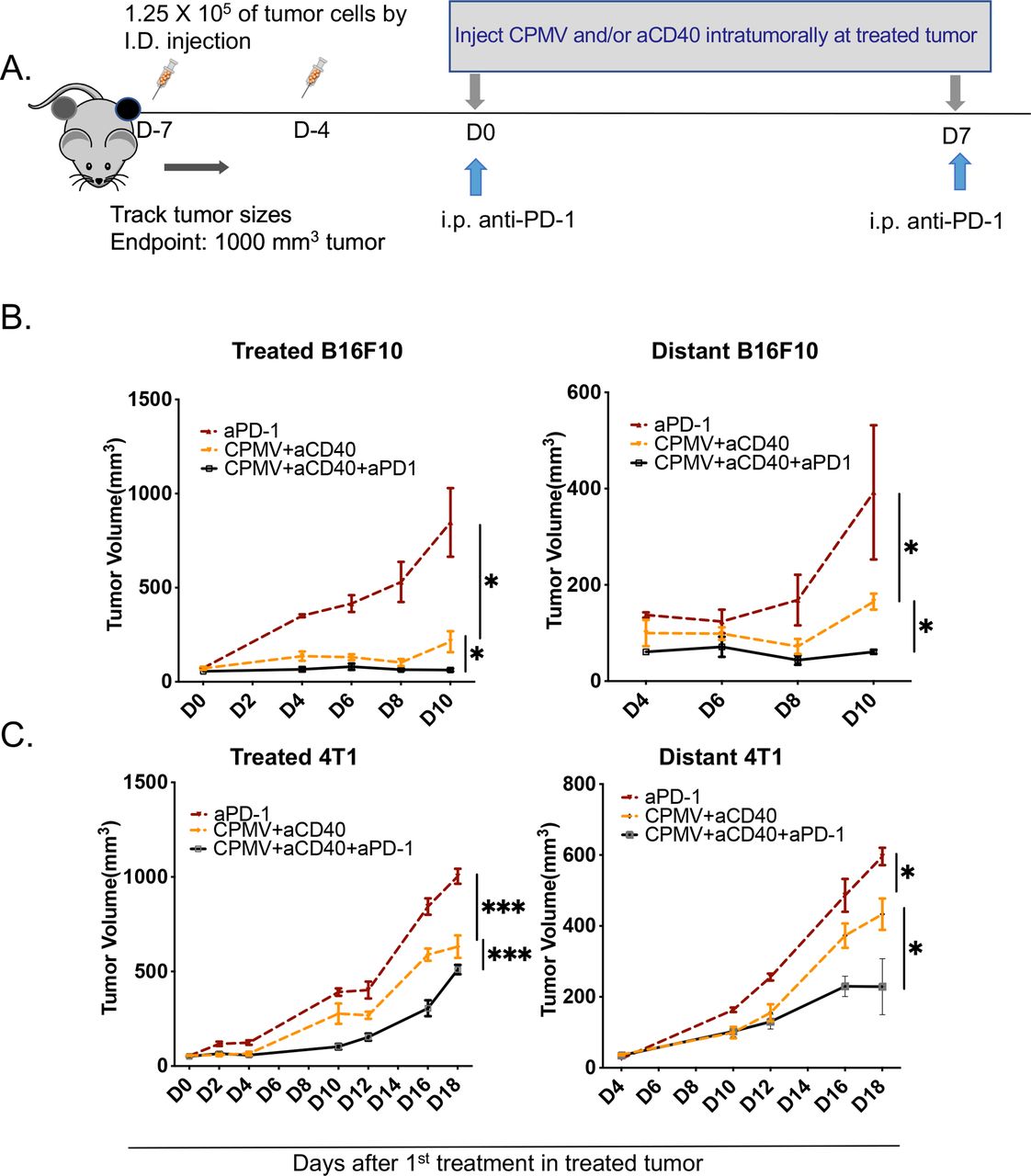 Figure 5
Figure 5 In situ vaccination combinations synergize with anti-PD-1 and elicit potent systemic antitumor immunity. (A) Schematic presentation of experimental setup. Two-tumor bearing mice of B16F10 or 4T1 had treated tumor injected weekly with CPMV and/or anti-CD40 and/or concurrent intraperitoneal injection of anti-PD-1. (B, C) Growth curves for corresponding tumors. Tumor growth curves were analyzed using two-way analysis of variance, with p>0.05 as ns, p<0.05 as *, p<0.01 as **, and p<0.001 as ***, p<0.0001 as ****. All experiments are repeated at least once and each with n≥3 mice/group. aCD40, anti-CD40; aPD-1, anti-PD-1; CPMV, cowpea mosaic virus; I.D., intradermal; i.p., intraperitoneal; PBS, phosphate buffered saline; PD-1, programmed death 1.
Serial ISV combinations overcome acquired resistance and elicit tumor specific memoryIt is well known that many immunotherapies fail due to acquired resistance at later time points after initial tumor response.13 We investigated whether serial ISV treatments combined with systemic aPD-1 therapy in the B16F10 model will dependably eliminate treated and untreated tumors using the timeline shown in figure 6A in which treatments were given every 5 days with different numbers of treatments in each group, (T as CPMV+aCD40+aPD-1; T×1 had one treatment, T×2 had two treatments etc). Continuing tumor regressions were seen after each cycle of ISVs in combination with aPD-1 therapy, resulting in durable responses in the treated mice after the fourth cycle and some complete eliminations of tumors on both flanks with localized vitiligo (figure 6B and online supplemental file 2) in B16F10 tumor-bearing mice. Surviving B16F10 mice rejected the same B16F10 tumors but not unrelated MC38 tumors, demonstrating tumor-specific systemic immunological memory (figure 6C; online supplemental file 3). Moreover, the combination had excellent efficacy and prolonged the survival in 4T1, in which severe necrotic tumors were observed even at day 60 after first treatment (figure 6D, data not shown). It should be noted that the 4T1 survival for the 4× treated mice was stopped due to this necrosis, which is assumed to be immune mediated; tumor regrowth was not evident. Taken together, these findings show that in situ induction and activation of adaptive immunity by cDC1s could overcome resistance toaPD-1 therapy and generate an abscopal effect that eliminated untreated tumors on the opposite flank.
 Figure 6
Figure 6 Extended in situ vaccination treatments overcome acquired resistance and elicit tumor specific memory. (A) Schematic presentation of experimental setup. Two-tumor bearing mice of B16F10 or 4T1 had the single-treated tumor injected every 5 days with intratumoral injection of PBS or CPMV and anti-CD40 and concurrent intraperitoneal injection of anti-PD-1 up to four times. (B) Tumor growth curves and survival curves for B16F10. (C) Tumor-free mice from (B) were rechallenged with B16F10 or MC 38 ninety days after first treatment in (B). (D) Tumor growth curves and survival curves for 4T1. Tumor growth curves were analyzed using two-way analysis of variance; Survival curves were compared using log-rank (Mantel-Cox) test, with p>0.05 as ns, p<0.05 as *, p<0.01 as **, and p<0.001 as ***. All experiments are repeated at least once and each with n≥3 mice/group. aCD40, anti-CD40; aPD-1, anti-PD-1; CPMV, cowpea mosaic virus; I.D., intradermal; i.p., intraperitoneally; PBS, phosphate buffered saline; PD-1, programmed death 1.
DiscussionOverall, our results demonstrate that CPMV, a multi-TLR agonist nanoparticle, stimulates potent systemic antitumor immunity when used as ISV. CPMV also works together with agonistic aCD40 antibodies as a powerful ISV doublet and reversed the resistance to aPD-1 in multiple ‘immune-cold’ mouse tumor models. These studies also illustrate the critical role of cDC1 for the new CPMV/aCD40 Ab ISV regimen to elicit CD8+ T-cell mediated tumor regressions of untreated tumors through cross-presentation. Moreover, repeated administrations of CPMV/aCD40 Ab ISV overcome the resistance to ICB, eliminated treated tumors and generated antigen-specific memory and robust systemic antitumor immunity that significantly suppressed often eliminated untreated tumors on the opposite flank. The studies illustrate the potential to generate a robust ‘abscopal effect’ to fight established metastatic disease after local in situ vaccination of a recognized tumor.
Tumor resident DCs are generally functionally immature and can contribute to tumor immune tolerance.41 42 Evidence showed that TLR ligands alone are not always capable of breaking peripheral CD8+ T-cell tolerance43; however, activation of CD40 on DCs increases the ability to overcome this tolerance.23 This provides the rationale to include CD40 agonist with CPMV to form a new dual ISV. Although systemic CD40 activation may cause autoimmune damage, the treatments here are local injection of CD40 agonist antibody. We did not conduct a detailed study to rule out toxicity associated with these treatments, but no discernable toxicity was observed in the treated animals apart from fur depigmentation due to vitiligo (online supplemental file 2). Previous studies have shown that various single TLR agonists work in synergy with aCD40 and to induce systemic antitumor efficacy.44 45 Our work with CPMV, a multi-TLR agonist nanoparticle, agrees with these findings. It will be of future interest to compare CPMV with some of the more popular single soluble TLR agonist in combination with CD40 agonist to determine whether the multi-TLR agonist and nanoparticle aspects of CPMV provide a unique ISV therapy option.
ICB is an established treatment approach that, despite considerable expense and common adverse events, helps a significant fraction of patients. However, common resistance to ICB, either primary or acquired, supports the need for additional combinatorial immunotherapy to extend benefits to more patients. Other studies have evaluated ISV to combat the common resistance to ICB, such as using papaya mosaic virus nanoparticles or TLR9 agonist virus-like particles for ISV46 47 and those and other studies support this possibility. Here, we demonstrate that cDC1 and CD8+ T cells are required for potent efficacy and abscopal effect of CPMV ISV; we also found that efficacy is significantly weaker without IFN-γ, or IL-12—as previously shown.6 This agrees with the finding that aPD-1 therapy depends on the interaction between CD8+ T cells and DCs through IFN-γ and IL-12.38 Moreover, with the trio combination of local CPMV plus aCD40 and systemic aPD-1, we observed eliminations of tumors after repeated administration at both treated and distant site, which is rare prior to this study.
We have also observed a general trend of better efficacy at treated tumors than the untreated tumor across different tumor models. The cause of the more robust tumor control at the treated site compared with the distant site, a finding with multiple in situ vaccines,48 49 is likely because the TME of the treated tumor is directly changed to be more immunostimulatory, while the impact on untreated tumors is from overall increases in systemic antitumor immunity. Of note, CPMV ISV does not require T cells to significantly slow tumor growth of most tumors directly treated with ISV. It appears that other phagocytic cells are primarily mediating the local efficacy induced by CPMV since phagocytes such as macrophages and neutrophils actively ingest CPMV and respond to TLR agonists.34 50 Importantly, the fundamental goal of ISV is to optimize systemic immunity to fight metastatic disease, whether recognized or not. The optimal way to use ISV to modify the TME of the treated tumor in order to generate a broadly effective systemic abscopal effect to eliminate untreated tumors remains unclear and is currently being investigated in our and other laboratories.
We recognize that the use of time-staggered bilateral implanted syngeneic tumor models as the induced metastatic models in this study does not fully reflect the multistep metastatic process in human patients.51 In our bilateral syngeneic tumor models, although we were able to recapitulate the distant seeding and colonization, we omitted the local invasion and intravasation steps. Moreover, we also limited the metastasis to the same tissue type, that is, there is also a lack of metastasis to multiple different organs. While this approach is able to document the abscopal effect of intratumoral treatment on a distant second tumor, it does not reflect the complex process of metastasis. Thus, future studies are warranted to assess this new combination for its antitumor ability in genetically engineered mouse tumor models.
Nonetheless, from a clinical perspective, the new treatment combination of CPMV/aCD40/aPD-1 still possesses significant therapeutic potential. The overall ISV strategy and specifically the use of CPMV for ISV has a variety of advantages for local and systemic solid tumor immunotherapy: (1) ISV uses low doses for intratumoral treatment relative to systemic immunotherapy. This lowers cost, reduces adverse events and as shown here and other studies, improves the response to the well-established options of ICB; (2) compared with other vaccination strategies, ISV circumvents the need to identify and synthesize neoantigens specific to individual patients’ tumors and in coordination with their human leukocyte antigen (HLA) alleles. The tumor itself, and all the recognizable antigens of any sort it contains, is used as the source of antigen for a therapeutic vaccine. This is a powerful way to bridge local and systemic therapies for better overall outcome. (3) ISV can be done rapidly and has rapid impact as shown here, so it could be applied to primary tumors as neoadjuvant therapy in the usual 10–21 days prior to surgical removal of a primary tumor. This would be an inexpensive and low-risk treatment to simulate systemic antitumor immunity when metastatic disease is a risk. (4) CPMV-ISV avoids limitations associated with pathogen-based (eg, viral and bacterial) approaches that require infection and can be blocked by neutralizing immunity, since pre-exposure to CPMV and the associated anti-CPMV immunoglobulin does not impair, and appears to improve, CPMV ISV.52 (5) By generating increased tumor specific effector/memory T cells, it is possible that systemic levels of ICB can be reduced which would reduce therapy-limiting off target autoimmune effects.
ConclusionIn summary, we have demonstrated significant abscopal effects from local ISV using CPMV alone. We also tested a dual ISV strategy of CPMV/aCD40 to activate cDC1s and effector T cells and data suggest this dual ISV to be safe, effective, and able to overcome resistance to systemic aPD-1. Systemic agonistic aCD40 Ab has been investigated previously24 and here CD40 agonist shows significant value as a locally administered in situ vaccine combined with CPMV and systemic PD-1 blockade. Our results demonstrate the trio combination of local CPMV/aCD40 and systemic aPD-1 is powerful and able to eliminate local and distant tumors after repeated administration. Our study also provides the rationale to include multi-TLR-agonists with DC agonists as local ISV.
A significant value of ISV is that, as shown here, even a short period of ISV can generate significant immune-mediated reduction in growth of untreated metastatic tumors. This rapid response to ISV, in association with speed of treatment delivery, expected moderate treatment cost, and excellent safety, supports the potential of ISV with CPMV during the roughly 10–21 days between pathological diagnosis and surgical removal of the primary tumor in most patients. Future studies will be needed to fully elucidate how other intratumoral therapies, including other TLR agonists, may impact T-cell exhaustion and therapeutic efficacy in distant tumors. Novel TLR9 agonists, for example, are thought to induce a systemic effect by activating plasmacytoid DCs (pDCs).53–55 Whether the pDCs trigger an immune response through direct antigen presentation versus the secretion of type 1 IFN alone and how this may impact T-cell exhaustion in distant tumors are questions of significant interest.
Data availability statementData are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information.
Ethics statementsPatient consent for publicationEthics approvalNot applicable.
AcknowledgmentsThis study was supported by Dartmouth Mouse Modeling Shared Resource, Dartmouth Irradiation, Preclinical Imaging and Microscopy Shared Resource and Dartlab Immune Monitoring Shared Resource, which receive support from the Dartmouth Cancer Center, through NCI funded grant 5P30CA023108. Graphic abstract is created by BioRender.com.
留言 (0)