Explanation for the choice of comparators
The summary description and intended purpose of the investigational device are the same as that of the comparator device. The investigational device differs from the comparator device in that its surface which has been modified by the addition of an antibacterial coating by integrating silver agglomerates using a proprietary Plasma Electrolytic Oxidation (PEO) process. Therefore, the comparator device will allow for the safety and clinical benefit of the modified surface to be assessed.
Intervention description
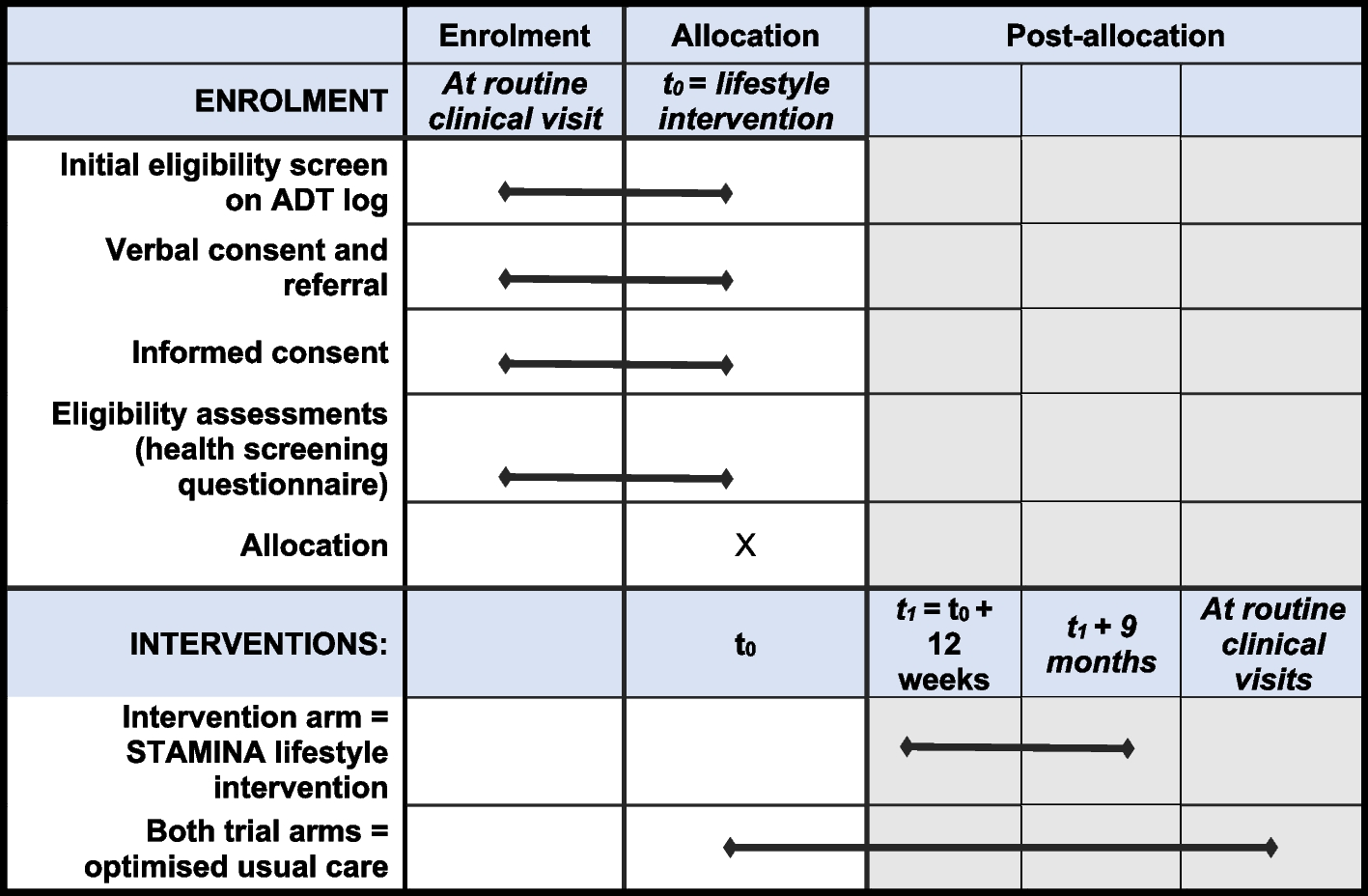
Regardless of whether the patient receives the Investigational Device or the Comparator, the study schedule is identical (see Table 2). Each patient will have seven study-related visits.
Screening and enrolment
After the patient gave consent, a preoperative baseline for all collected assessments in connection with the defined endpoints will be done. Attention must be paid to the baseline concentration of silver in the blood, since external factors (i.e., working in a photo development laboratory) can have an influence that could falsify the study results if this is not taken into account.
Implantation
On the day of the implantation the patient will be randomized to one of the two treatment groups. No specifications are made by the clinical investigation plan for implantation and subsequent care. This lies in the assessment of the treating physician. The first sample for silver level analysis after implantation must be done 24 h (± 12 h) after wound closure. If a drainage has been applied to the patient, a sample of the wound fluid will be collected 24 h (± 12 h) and 48 h (± 12 h) after wound closure to be able to compare the expected high silver concentration on the local area around the implant to the silver concentration in the blood.
Follow-up Visits
At the five follow-up (FU) visits of up to a year the patient will be accompanied in his or her healing process and all assessments for the defined endpoints will be made. The point of study termination is defined as the date of 12-month FU for patients with regular study termination. Following completion of the 12-month FU visit, the subject will be treated according to the standard of care practice of the treating surgeon.
Criteria for discontinuing or modifying allocated interventions
At any time and without any reason, patients can withdraw from the clinical trial without consequences. Because of compliance reasons, the investigator can end the patient’s participation. The collected data of patients who did not end the trial per protocol will be part of the Safety Analysis Set.
Strategies to improve adherence to interventions
Surgeons will be trained in handling of the implants to avoid any issues with the wrong use of the medical device. The study sites will be closely monitored on-site and remote by a clinical research associate (CRA). The CRA will check the training records of the study team and verify the source data. In the course of the clinical trial, an unblinded CRA will visit the study site for implant accountability, counting the implant packaging, and reviewing the device deliveries.
Relevant concomitant care permitted or prohibited during the trial
No concomitant care and interventions are prohibited during the trial.
Provisions for post-trial care
Following completion of the 12-month FU visit, the subject will be treated according to the standard of care practice of the treating surgeon. The Sponsor will maintain an adequate insurance policy covering damages arising out of the clinical trial. This insurance covers the subjects with respect to the risks involved in this study according to the clinical investigational plan.
Outcomes
Since this clinical trial should demonstrate the non-inferiority of the silver-coated implants the primary outcome is the comparison of predefined AADEs of patients treated with the investigational product and patients who received the comparator. A list of AADEs was defined based on experiences with the non-coated implants (see Table 1). Other outcomes of interest are:
To investigate the proportion of subjects with device-related infections occurring after successful implantation of the study device and end of the 12-month FU and compare the rate between the treatment arms.
To investigate fracture healing assessed by local and central reviewer and compare the rate of completely healed subjects between treatment arms.
To investigate the number of hospitalizations occurring in the first 12 months post-implantation and the nights spent in hospital and compare the numbers between treatment arms.
To investigate the change in American Orthopaedic Foot and Ankle Score (AOFAS) at each FU visit and compare endpoint between the treatment arms.
To investigate the change in average pain at rest at each FU visit and compare endpoint between the treatment arms.
To investigate the change in the disability rating index at each FU visit and compare endpoint between the treatment arms.
To investigate all items assessed in the EQ-5D-5L questionnaire.
To investigate the proportion of subjects with full weight bearing at each FU visit and compare this endpoint between the treatment arms.
To investigate the change in silver serum levels at each scheduled FU visit and compare endpoint between the treatment arms. The change in silver serum level is defined as the difference between the respective silver level at the respective FU and the silver level at Screening/Enrollment Visit.
To investigate the proportion of subjects with Treatment Emergent Adverse Events (TEAEs) during the 12-month FU and compare the rate between treatment arms. A TEAE is considered as any AE observed during the 12-month FU which occurred after the start of the implantation surgery.
Table 1 List of anticipated adverse device effectsParticipant timeline
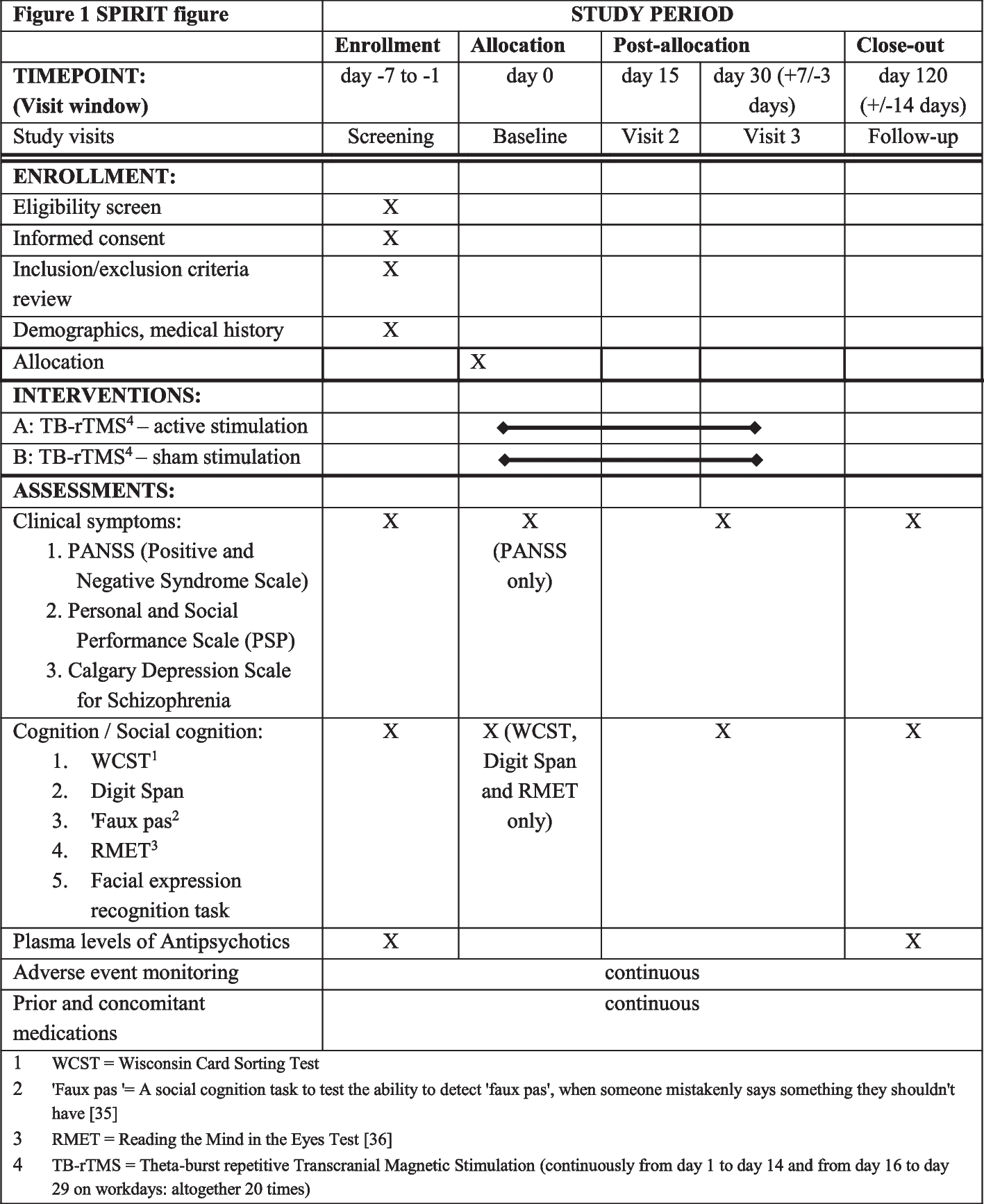
The participant timeline is shown in Table 2.
Table 2 Study schedule. Assessment windows will be as follows: screening/enrollment visit (day −21 to day 0), implantation (day 0), 1-week FU visit (7 ± 3 days), 6-week FU (42 ± 7 days), 3-month FU (90 ± 14 days), 6-month FU (182 ± 14 days), 12-month FU (365 ± 30 days)Sample size
The sample size calculation is based on the number of expected AADE of uncoated system compared to the coated system. A one-sided test at the 5% significance level and 80% power requires 96 subjects successfully implanted with either system to detect non-inferiority at a margin of 10%, i.e. an overall number of 192 subjects with successfully implanted study devices. Accounting for a 15% drop–out rate due to screening failures and surgical failures a total sample size of 226 screened subjects is considered sufficient to achieve the primary objective of the study.
Recruitment
Patients with fractures of the distal tibia will be recruited at up to 20 German study sites.


留言 (0)