
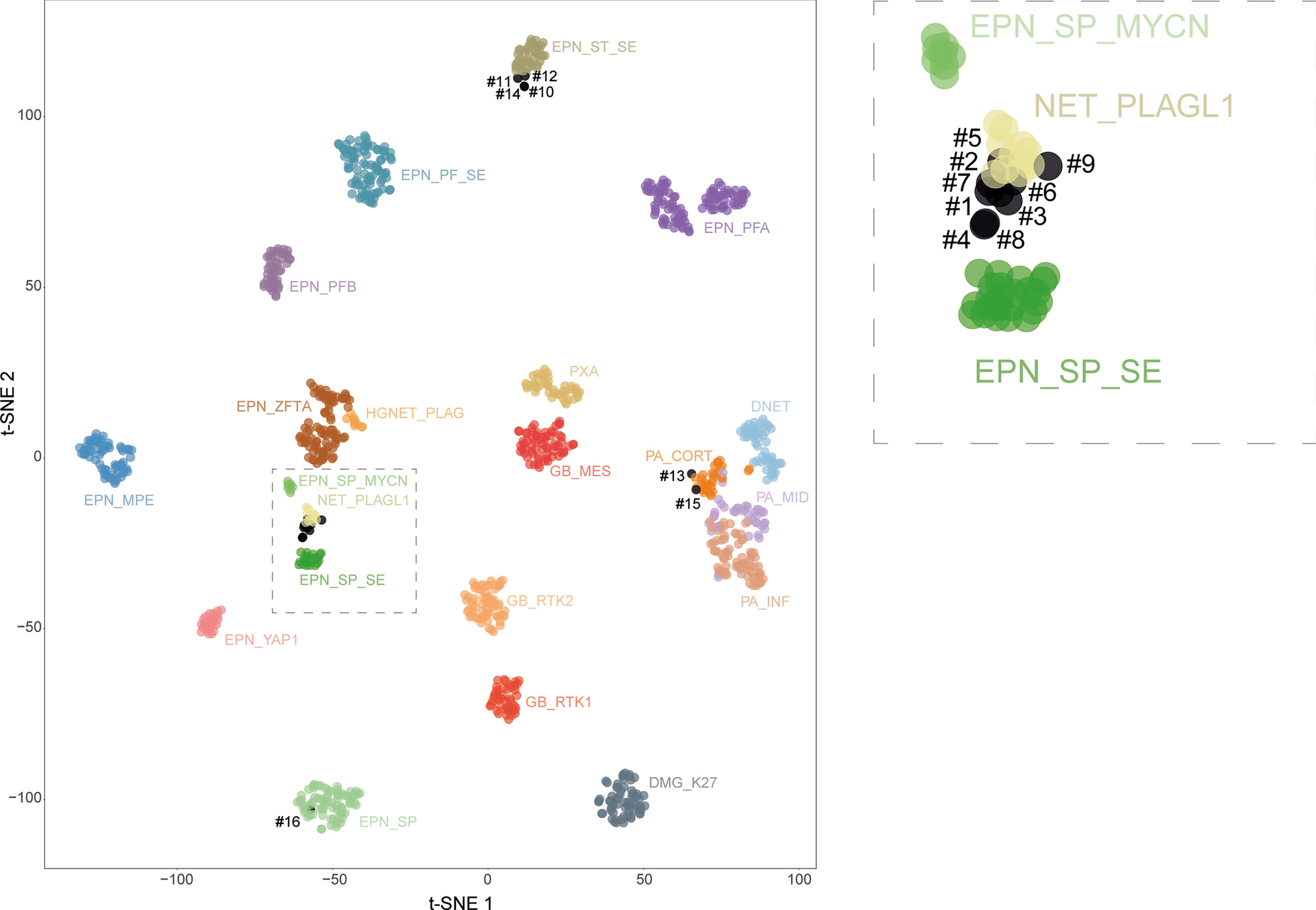
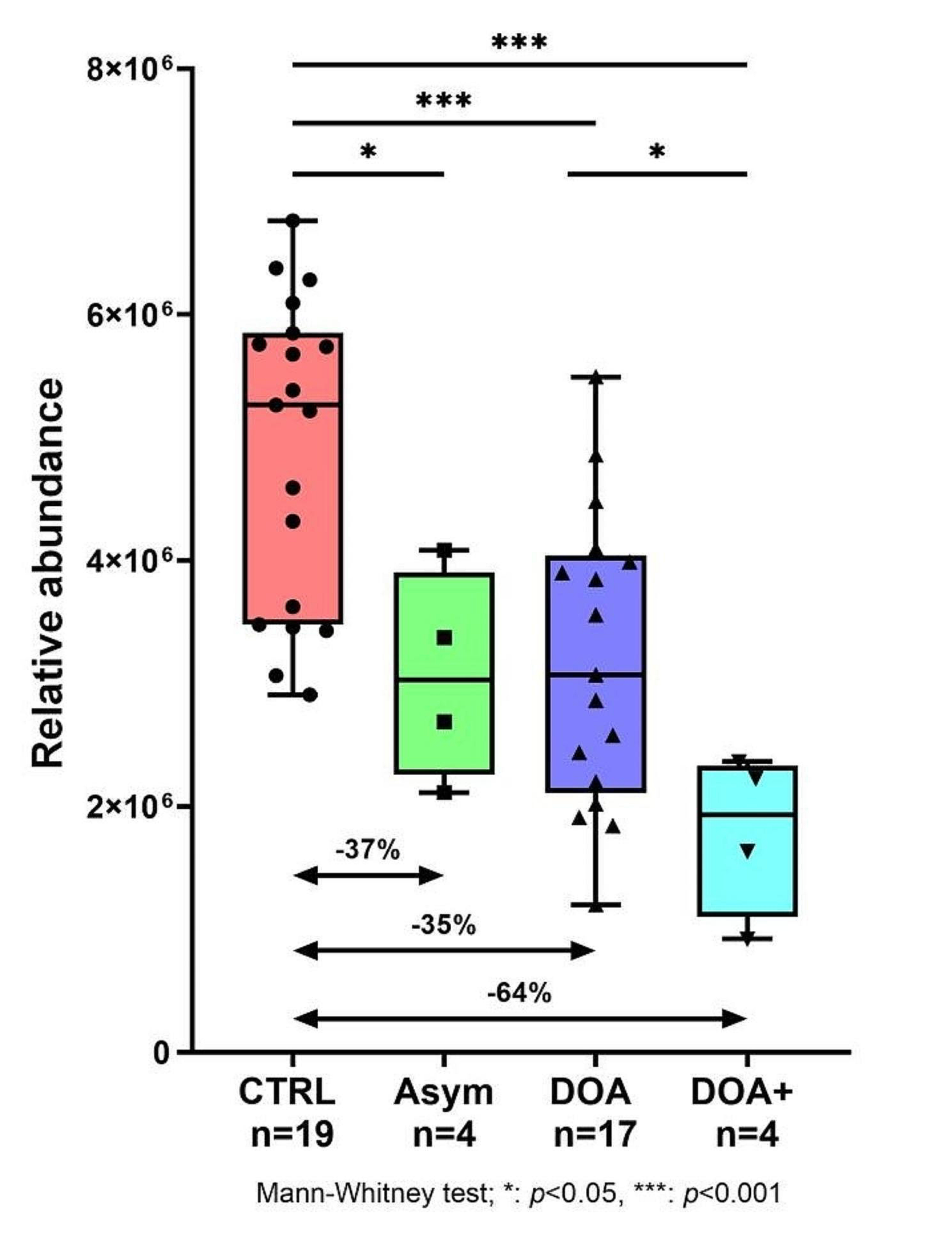
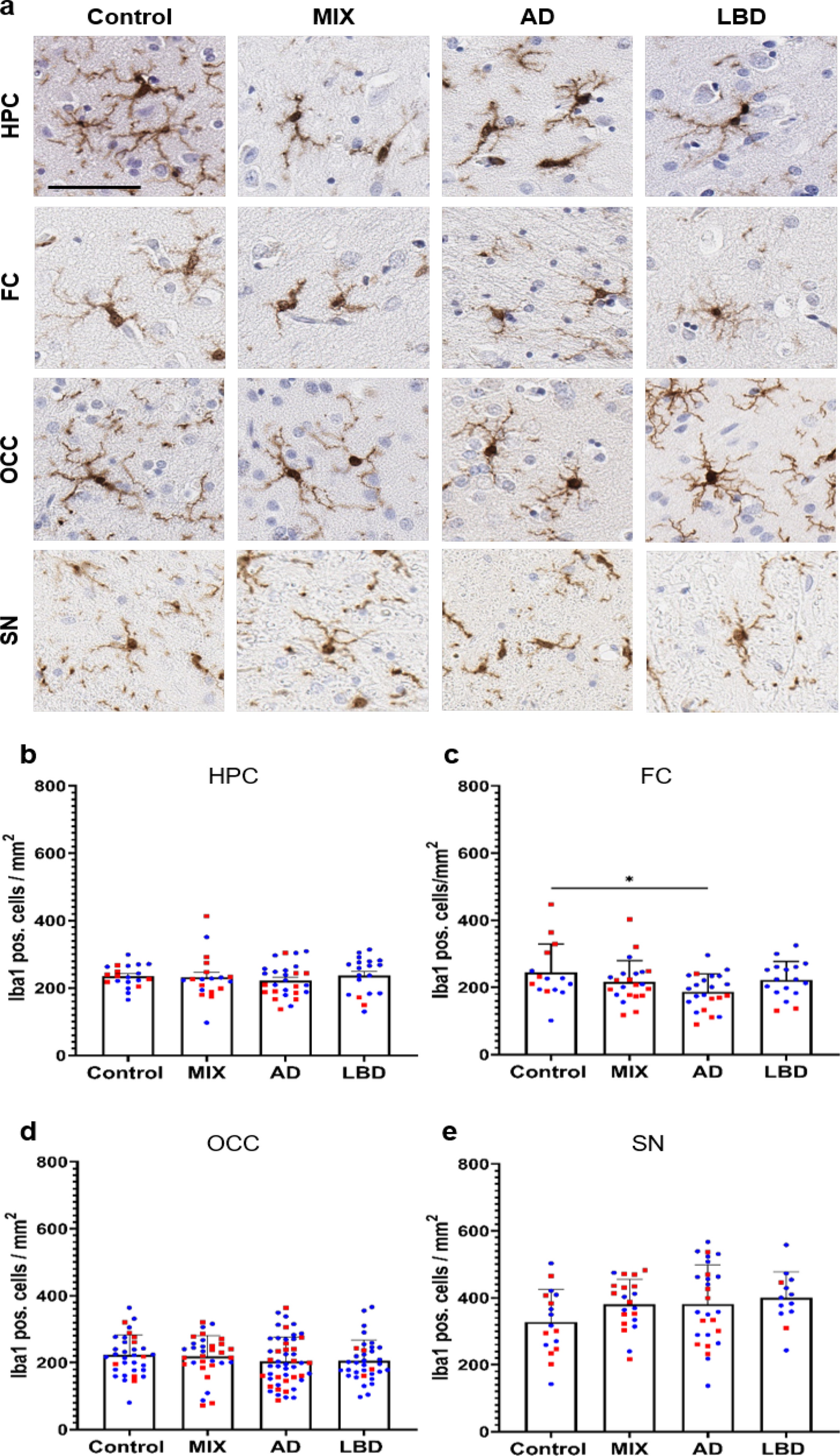
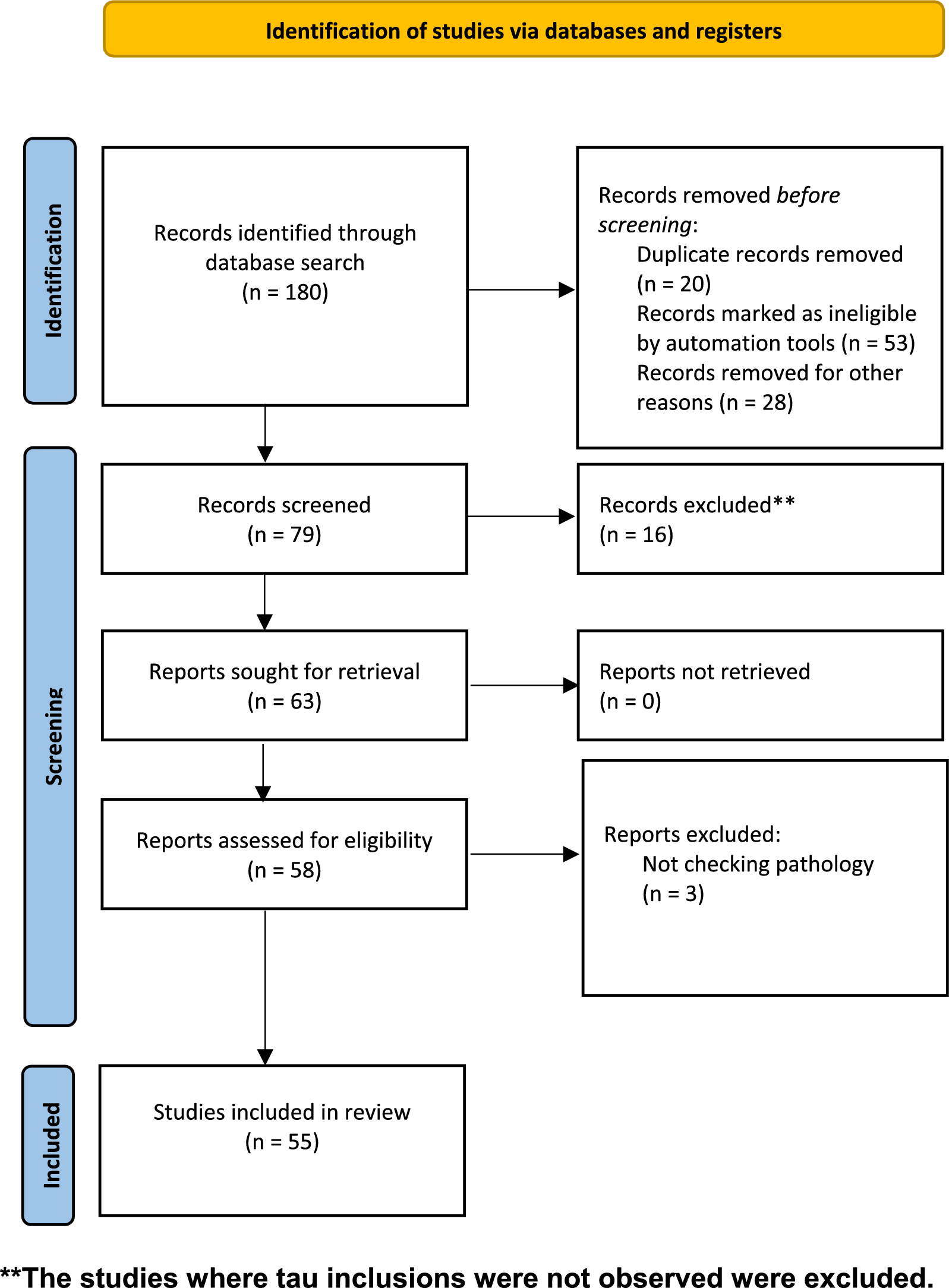
記住我
We used scWGS to determine the frequency of CNVs in the temporal cortex, hippocampal subfields cornu ammonis (CA) 1, hippocampal subfields cornu ammonis (CA) 3, cerebellum (CB) and entorhinal cortex (EC) of 13 AD patients and 7 age-matched healthy controls (Figs. 1A, 2A,B, Additional file 1: Table S1). The Braak stages of AD patients ranged between III and VI (Fig. 2C). Neuronal nuclei were isolated using either FACS (sorted with propidium iodide, n = 12) or LCM (sorted with cresyl violet, n = 1552), the latter performed on frozen brain slices (Fig. 1B, see Methods). LCM-isolated non-neuronal “blank” regions were used as negative control (n = 10). The LCM method, although more difficult to implement than FACS, was chosen to ensure the selection of nuclei of pyramidal neurons for sequencing, known to be particularly sensitive to AD [21]. For technical comparison, neurons of a single individual were collected both using FACS (n = 12) and LCM (n = 64) (see Methods). scWGS libraries were prepared using GenomePlex whole-genome amplification and specific adapters were inserted using Phusion® PCR. Paired-end reads were mapped to the human reference genome, followed by stringent filtering to obtain uniquely mapped reads (see Methods). This resulted in a median of 276,446 reads, corresponding to a coverage of 0.006X per LCM-isolated cell (range [133–1,909,016] reads and [0.000003X–0.04X] coverage) (Fig. 3A).
Fig. 1
Schematic of the workflow and information about the samples. A The pipeline of NGS data analysis and CNV detection. B Images from a frozen hippocampal brain slice stained with cresyl-violet showing a pyramidal cell before (B1) and after (B2) laser capture microdissection-based isolation process using the PALM device. Circles in B1 indicate positions where two pyramidal cells have already been isolated just prior to the picture being taken. Scale bar, 50 µm
Fig. 2
Sample information. A Bar plot showing the number of cells that have been sequenced for each individual. Brain regions are illustrated in different colors (see the colour key on the top of the figure). B Dot plot showing age of AD (pink) and control (green) individuals. C The table summarises sex, diagnoses and Braak level of the individuals. Detailed information on the individuals is available at Additional file 1: Table S1-A
Fig. 3
Comparison between different cell isolation methods and published datasets. Box plots showing the distribution of coverage (A), mapping proportion (B), index of dispersion (C) among FACS-isolated, LCM-isolated and LCM-isolated blank samples. P-values were calculated using Kruskal–Wallis test among groups and Wilcoxon rank-sum test between groups. (D) Violin plot showing the distribution of coverage among different datasets. P-values were calculated using Wilcoxon rank-sum test between groups. This study, including only LCM: blue; van den Bos 2016: brown; McConnell 2013: purple. (E) A principal components analysis (PCA) was performed using the normalized read counts across autosomal bins (n = 5243) in published datasets and this study. Because they dominated the PCs, cells deviating from the [1.9–2] range were not included in the analyses. The number of cells for each dataset are indicated on the plot. X-axes illustrate PC1 and PC3 that explain 18.4% and 1.3% of the total variance, respectively. Y-axes show PC2 and PC4 that explain 2.8% and 0.9% of the total variance, respectively. (F) Boxplots showing the distribution of median CN of chromosome 1 (chr1, upper part of the figure) and chromosome 21 (chr21, lower part of the figure) across bins (n = 440 and n = 68 for chr1 and chr21, respectively). Each point corresponds to the median CN of each cell. Minimum (“Min”), median (“Med”), maximum (“Max”) and standard deviation (“sd”) of each distribution were shown on the boxplot. Cells that deviated from the [1.9–2] range were excluded from the analyses to be consistent with our filtering criteria (except for the uncorrected datasets). This study [Uncorrected (n = 1337), Uncorrected-filtered (n = 588), PCA-corrected (n = 1301)]: blue; van den Bos 2016 (n = 1468): brown; McConnell 2013 (n = 109): purple
CNVs were predicted using Ginkgo, which uses circular binary segmentation (CBS) to estimate deletion or duplication events [45]. Negative controls (n = 10) and FACS-isolated neurons (n = 12) were analyzed separately and are not included in the main results. Ginkgo was run on our dataset with n = 1542 cells, while in parallel, we also analyzed two published scWGS datasets: one by van den Bos and colleagues (“van den Bos 2016”), comprising n = 1469 cells from healthy and AD brains (median coverage 0.005X) and another by McConnell and colleagues (“McConnell 2013”), comprising n = 110 cells from healthy brains (median coverage 0.047X) (Fig. 3D) [5, 48]. Note that the van den Bos 2016 dataset includes only 61% of cells produced in that study, because data from cells filtered for high noise levels were not published and thus could not be included here.
LCM-isolated cells show a high frequency of depth variabilityWe first evaluated the sensitivity and specificity of our bioinformatics pipeline on scWGS data using trisomy-21 in DS and monosomy-X in males in published data. Analyzing n = 34 neuronal nuclei from DS individuals [5], trisomy-21 was correctly predicted across all samples without any false positive or false negative calls. In addition, monosomy-X was accurately predicted in 94.2% (338 of 359) of cells from males across the two published datasets [5, 48].
Ginkgo includes an algorithm that uses the distribution of read depth across the genome to infer the average DNA copy number of each cell, which is estimated within a range of 1.5 to 6. It would be expected that the majority of human neurons would carry on average two copies of each autosome, although high frequencies (10–35%) of hyperploid neurons have also been reported, especially in AD brains [10].
Applying Ginkgo on the two published datasets, we found that for 99.9% (1577 of 1579) of cells the estimated average copy number lies within [1.9–2]. Using the same algorithm on our dataset, however, only 45% (687 of 1542) of the cells had average copy numbers estimated within the [1.9–2] range; i.e. 55% were non-euploid. Although hyperploid neurons have been described in control brains at ~ 10% frequency using FISH [10], the observed non-euploidy estimates suggest that our dataset carries particularly high levels of variability in read depth. These differences, in turn, could be related to the LCM protocol used, as the published scWGS experiments had used FACS.
To investigate this possibility, we compared the quality metrics of cells we had collected using FACS or LCM for this study. These metrics were mapping proportion (the number of mapped reads/ the total number of reads), coverage, and index of dispersion (IOD, the ratio between the variance of read coverage and the mean). FACS-isolated cells had higher sequencing coverage and mapping proportions than the LCM-isolated ones (Wilcoxon two-sided rank-sum test, p < 0.0001 and p < 0.001 for coverage and mapping proportion, respectively) (Fig. 3A, B). Note that the difference in coverage variability between FACS and LCM has not been reported elsewhere. In addition, FACS-isolated cells had low IOD values, indicating less variation in sequence depth than the rest of the samples (Kruskal–Wallis test, p = 1.5e−07) (Fig. 3C). Because our LCM and FACS samples originated from different brain regions with different cell type proportions, we also asked whether such differences could explain the observed LCM vs. FACS differences. To rule out this possibility, we compared the index of dispersion value of the cells that were taken from the temporal cortex of the same individual using FACS (n = 12) and using LCM (n = 64). We found a significant difference in the direction of higher variability in LCM (Wilcoxon rank-sum test p < 0.001), indicating that the observed variability between LCM and FACS can not be simply explained by differences in cell type proportion among brain regions. We note that the higher noise observed in LCM data was not solely due to higher genome coverage, as the FACS-based data from the van den Bos 2016 dataset had a median coverage comparable to ours (0.005X vs. 0.006X), but did not show comparable variability as in our LCM data (Fig. 3D). These differences in IOD between LCM and FACS could be potentially explained by the higher sensitivity of the LCM procedure to experimental noise, compared to FACS. Alternatively, they could partly represent abnormal nuclei selected out in FACS but captured by LCM.
We next investigated the possibility that underlying variation may be caused by technical and/or biological factors. For this, we used a generalized linear mixed model (GLMM) to explain IOD (the response variable) per LCM-isolated cell (n = 1542) as a function of diagnosis (AD vs. control), genome coverage, and brain region as fixed factors, and individual as a random factor (see Methods). Note that p-values of the pairwise differences between AD and control (Fig. 4A–C) was calculated using a linear mixed-effects model (see Fig. 4 legend). We found that coverage has a significant negative effect on IOD, as may be expected (z = -21.06, p < 0.0001). Compared to the cerebellum, the region least affected by neurodegenerative diseases [34], we found a significantly high IOD for the entorhinal cortex (z = 2.61, p < 0.05), hippocampal CA1 (z = 3.34, p < 0.001) and hippocampal CA3 (z = 3.75, p < 0.001), but not for the temporal cortex (z = − 0.28, p = 0.78) (Fig. 4B). Finally, neurons from control individuals have slightly less IOD than AD patients (z = − 1.93, p = 0.054) (Fig. 4A–C). This result might suggest a tendency for neurons of AD patients to carry more variable DNA content and is consistent with cytometry analyses reporting a high occurrence of hyperploid neurons in the AD brain [10]. Although these findings imply a role of biological factors in read count variation within cells, it still remains possible that confounding technical factors influence our data. Given this uncertainty about the source of variability, we continued the analyses by filtering our dataset to remove the most variable cells.
Fig. 4
The distribution of index of dispersion (“IOD”) for LCM-isolated cells (n = 1542) according to A diagnoses (Alzheimer’s disease (“AD”), control), B brain regions (hippocampal subfields CA1 (“Hippocampal CA1”), hippocampal subfields CA3 (“Hippocampal CA3”), Entorhinal cortex, Cerebellum, Temporal cortex). For each brain region, we tested whether AD diagnosis was predictive of IOD using a linear mixed-effects (lme) model (see Methods). Individuals were added as a random factor. Across all tested brain regions, differences were only marginally significant (p = 0.069). C The distribution of IOD across individuals (n = 20). Box plots were ordered by the median. Y-axes illustrate the IOD values on the log10 scale
No significant difference in CNV frequency between AD and control in the “uncorrected-filtered” datasetWe then used Ginkgo to call CNV events from the "uncorrected-filtered" dataset (n = 882 cells from 13 AD patients, and n = 660 cells from 7 healthy controls). We found 19,608 events in 882 cells from AD patients (22.2 per cell), and 14,844 events in 660 cells from healthy controls (22.5 per cell). We then tested the observed frequency difference between AD and control using a GLMM with a negative binomial error distribution (see Methods). The response variable (the frequency of CNVs) was predicted using a combination of fixed factors, including diagnoses, chromosomes, brain regions, sex and coverage (Fig. 6D). The individual effect was added as a random factor. We found no statistically significant difference between AD and control across all tested combinations (GLMM, p ≥ 0.17; Additional file 3: Table S3).
CNV estimation from low coverage scWGS data is known to be highly sensitive to technical noise, and a large proportion of the called CNV events likely represent false positives. We thus decided to filter both cells and CNV events in our dataset to obtain a more reliable dataset [6, 52, 53]. We started by removing the most highly variable cells among the LCM-isolated ones (n = 1542) using the following criteria. First, 13% (205 of 1542) of the cells with a low number of reads (< 50,000) were discarded from the analysis (see Methods). Second, as most cells are expected to be diploid, and also given that the Ginkgo-estimated copy number (CN) profiles of 99% of cells in the McConnell 2013 and van den Bos 2016 datasets were observed to lie between [1.9–2], we excluded those cells with CN values beyond this range (54% excluded, 726 of 1337). Third, we filtered out 23 of the remaining 611 cells (4%) that showed extreme CNV intensity, which we defined as three or more chromosomes of a cell carrying predicted CNVs that cover > 70% of their length (Fig. 6A). Information about the remaining cells (n = 588) is provided in Additional file 2: Table S2 and Additional file 4: Fig. S1.
From these 588 cells, we called 3521 CNVs (~ 5.9 events per cell) in the uncorrected data, which we call the “uncorrected-filtered” dataset. We further applied a number of conservative filtering criteria to remove potential false positives: (1) We only included megabase scale CNVs (≥ 10 Mb), considering that detection of small events with low coverage data will be unreliable. (2) We limited the analyses to 1-somy and 3-somy events, assuming that most somatic CNVs involving chromosomes or chromosome segments would involve loss or duplication of a single copy. (3) We only included CNVs with unique boundaries across all analysed cells, assuming that somatic CNV breakpoint boundaries should be generally randomly distributed across the human genome. (4) We removed CNVs on the proximal portion of the chr19 p-arm, where frequently observed duplications were previously reported as low coverage sequencing artifacts [43]. (5) To ensure the reliability of the CNV signal, we calculated a standard Z-score for each CNV that reflects the deviation in read count distribution in that region compared to the rest of the cell (which we call Z1, see Methods), and only accepted CNVs with absolute values of Z1-scores ≥ 2. (6) We reasoned that read counts in a real CNV should be closely clustered around expected integer values (e.g. 1 or 3). To assess this, we calculated a Z-score for the deviation from the expectation (called Z2), and only accepted events with absolute values of Z2-scores ≤ 0.5 (see Methods, Fig. 6A, Additional file 4: Fig. S3).
After CNV filtering, we found 12 CNV events across 295 cells in 13 AD individuals and 4 CNV events across 293 cells in 7 controls. Among the 295 pyramidal neurons analyzed from the 13 AD patients, we found 10 deletions (3.39% per cell) and 2 duplications (0.68% per cell) (Fig. 6B). These events ranged in size from about 10.14 to 77.01 Mb (median: 19.31 Mb) and were observed in the temporal cortex and the entorhinal cortex. Of the 293 neurons from 7 control brains, 1 deletion (0.34% per cell) and 3 duplications (1.02% per cell) were detected in the temporal cortex with a size range of 10.81 to 54.67 Mb (median: 14.51 Mb) (Fig. 6B). Again testing the CNV frequency differences between AD and control brains using a GLMM, we found no statistically significant effect (GLMM, p ≥ 0.88) (Additional file 2: Table S2, Additional file 3: Table S3).
We also implemented an alternative algorithm, HMMcopy [46], to predict CNVs (see Methods). Overall, 75% (12/16) of the HMMcopy predictions overlapped with the CNV events that we found after filtering the uncorrected Ginkgo predictions. Comparing predicted CNV event frequencies between AD and control we again found no significant difference (z = -1.34, p = 0.18).
A PCA-based denoising approach minimizes within-cell depth variabilityTo gain further insight into within-cell variability in our dataset (the uncorrected-filtered version) compared to the two published scWGS datasets, we calculated the median CN of chr1 and chr21 (the largest and smallest chromosomes) across all three. We still found conspicuously higher within-cell variation in our dataset, despite having discarded highly variable cells (Fig. 3F). We then used the autosomal normalized read counts to perform a PCA on the uncorrected-filtered data and published datasets. We also included blank (negative control) samples and FACS-isolated cells to illustrate how reads counts from these two groups relate to others. According to the PCA, LCM-isolated uncorrected-filtered data and blank samples were separated from the published datasets and FACS-isolated cells (Fig. 3E). This result might also highlight distinct profiles of LCM-isolated cells.
We then sought an approach that could reduce this elevated within-cell variability in read depth, assuming it is of technical origin and possibly related to the LCM procedure. Experimental biases could involve cross-contamination across cells during isolation, or biases that arise during DNA amplification. Although the former should be mainly random, the latter may follow systematic patterns, such as some chromosome segments being more or less prone to be amplified.
We thus devised a procedure for removing putative patterns of systematic read depth variation across cells (see Methods). The algorithm starts by choosing a focal cell x in the dataset, and calculating principal components (PCs) from the normalized read counts per autosome across the rest of the cells (except cell x). It then collects all PCs explaining ≥ 90% of the variance. Treating these as representatives of systematic variation, it removes their values from the normalized read counts of cell x using multiple regression analysis. These steps are performed on all cells individually, creating a “denoised” dataset. The final dataset contains residuals from the multiple regressions instead of the normalized read counts. Notably, this procedure should remove experimentally-induced variation in read depth shared among cells, and also any recurrently occurring somatic CNVs. Rare somatic CNVs, instead, would be mostly unique to each cell and randomly distributed in the genome, and thus would not be affected.
After filtering cells with a low number of reads (n = 205) and denoising our dataset with this approach, CN and CNV prediction were performed using Ginkgo. We further compared the results between the PCA-corrected and “uncorrected-filtered” datasets. Examples of cells having “noisy” profiles before and after correction are shown in Fig. 5A–C, which suggests a dramatic reduction in within-cell variability. Beyond visual inspection, we also analyzed three statistics. First, we studied the CN profile of cells after PCA correction. We found 97% (1302 of 1337) now lie between 1.9 and 2 (Fig. 6C). This result is comparable to the two published datasets described above and much higher than uncorrected data (45%). Second, we calculated the number of CNV events per cell (sum of the number of CNV/ number of cells) across datasets. In the van den Bos 2016 and McConnell 2013 datasets, we estimated 5.6 and 8.1 CNVs per cell, respectively (Fig. 5E). In our dataset, in the uncorrected version, we found 23.9 CNVs per cell, in the “uncorrected-filtered” data 6.0 CNVs per cell, and in the PCA-corrected data, we estimated on average 1.0 CNV event per cell. The denoising leads to lower CNV estimates in our data, which is more conservative and possibly more realistic than the higher estimates without correction. Third, we estimated the standard deviation in CN among cells for chr1 and chr21. For chr1 and chr21, the standard deviations in the PCA-based data were 4 and 2.3 times lower than in the “uncorrected-filtered” data, respectively, and comparable to CN standard deviations in the two published datasets.
Fig. 5
A PCA-based denoising approach helps eliminate technical noise. A–C Examples of CN estimates of cells using uncorrected data (upper panels) and using data after PCA-based correction (lower panels). The x-axes show chromosomes and the y-axes show the CN profile of chromosomes estimated by Ginkgo. Each grey dot represents the scaled and normalized read counts per bin. Amplifications (CN > 2) are shown in red; deletions (CN < 2) in blue; disomy (CN = 2) in black. Our denoising approach increased variability for only three cells tested (out of 1301), the CN profiles of which are shown in Additional file 4: Fig. S2. D Distributions of the number of CN before (n = 3,521) and after correction (n = 1,298) across autosomes. For illustration purposes, the bar plot includes up to 10-somy. E Bar plot showing the number of CNVs per cell across datasets. The datasets from our study include cells from both AD and control (Uncorrected, Uncorrected-filtered, PCA-corrected): blue; van den Bos 2016 (including cells from both AD and control): brown; McConnell 2013: purple). The number of cells that were used to calculate CNVs per cell was shown on the X-axis label
Fig. 6
Genome-wide distribution of predicted CNVs in neurons. A Overview of cell and CNVs elimination steps in the uncorrected and corrected data. We applied the same criteria for excluding CNVs to the PCA-corrected results. *We discarded CNVs if their breakpoints were within the three base pairs window around each other. B Bar plots represent the number of CNVs per cell for AD and control groups. Blue area: 1-somy (deletions); pink area: 3-somy (duplications). The number of unique individuals with at least one cell identified with a reliable CNV is indicated within the panel. C The table shows the ploidy levels of cells in corrected and uncorrected versions of the data. D The table shows several combinations of fixed factors that were used to test the difference between AD and control in the frequency of CNVs (“Freq of CNVs”). The fixed factors of the model included: diagnoses (AD and control), chromosomes (“Chr”, 22 autosomes as categorical variable), sex (male and female), brain regions (“Brain reg”, temporal cortex, hippocampus CA1, hippocampus CA3, cerebellum, entorhinal cortex) and coverage. Considering correlation due to repeated measurements on the same subject, individual (“Indiv”) effects were added as a random factor. In addition to the interaction of chromosomes and diagnoses (i.e. “Chr * Diagnosis + Coverage”), the effects of the chromosomes were also tested individually (i.e. “Chr + Diagnosis + Coverage”. The results were not significant across any of the seven GLMM models tested, in any of the analyses datasets. In the main text, we report the lowest “diagnoses” p-value across the seven tests. Note that we did not apply any multiple testing correction. E The heatmap shows the genome-wide copy number profile of cells analyzed in the corrected data (n = 10) with at least one reliable CNV. CNVs, brain regions and diagnoses are illustrated in different colors (see the color key on the left of the figure). Each row shows a cell and each column shows a chromosome
Subchromosomal CNVs are enriched in deletions in the PCA-corrected dataBased on these three statistics, we decided to study this PCA-corrected version of our dataset. For downstream analysis, we further eliminated cells that deviated from the ploidy range of [1.9–2] (2.6%, 35 of 1337) or showed extreme CNV intensity (0.08%, 1 of 1302) (Fig. 6A). We thus created a denoised dataset of 1301 pyramidal neurons from 20 individuals.
Estimating CNVs in this dataset using Ginkgo, we found 1298 CNVs in total (~ 1 event per cell). To remove false positives, we also performed the same CNV prediction and downstream analyses on our PCA-corrected data (Additional file 4: Fig. S3). After these steps, we found a total of 9 deletion events (0.7% per cell) and 1 duplication event (0.08% per cell) across 1301 cells in 20 individuals among all tested brain regions (except for the hippocampal CA1 where no CNV event was found). This excess of deletions is unexpected under the null hypothesis of equal expectation of duplication and deletions (two-sided binomial test p = 0.021), but consistent with previous observations of more deletions than duplications among somatic mutations [6, 48, 50, 53].
No significant difference between AD and control after PCA-correction or in the van den Bos datasetStudying CNV frequencies with respect to diagnosis, we found 6 CNV events across 688 cells in 13 AD individuals and 4 CNV events across 613 cells in 7 controls (Fig. 6E). Performing the formal test for the hypothesis of AD versus control differences with this data, we again found no significant difference between the groups (GLMM, p ≥ 0.80; Additional file 3: Table S3). Information about the CNVs and cells can be found in Additional file 2: Table S2.
We also repeated the same analysis on the van den Bos 2016 dataset, from which originally only aneuploidy was reported. Here we identified 11 CNV events across 883 cells in 10 AD individuals and 3 CNV events across 585 cells in 6 controls. The difference was in the same direction as in our dataset, but again not significant (GLMM, p ≥ 0.79) (Fig. 6B, Additional file 2: Table S2, Additional file 3: Table S3).
留言 (0)