Study setting
The study is carried out in DRC where malaria transmission is intense and perennial.
The study is conducted in the “Maternité Esengo,” a 106-bed health facility, located in Kisenso, Kinshasa, where there is an average of 100 deliveries per month. A previous study performed in a similar setting showed that the prevalence of malaria in pregnant women was 30% [4].
Pregnant women will be recruited during ANC visits, regardless of their parity.
Eligibility criteria
Inclusion criteria (all should be present):
1.
Gestation ≥16 weeks
2.
Age: ≥18 years
3.
Residence within the health facility catchment area
4.
Willingness to adhere to study requirements and to deliver at the health facility
5.
Willingness to provide written informed consent; if the woman is illiterate, she can choose an impartial witness, not related to the study, to accompany her during the consent process and they will both sign the informed consent form
Exclusion criteria:
1.
Known history of allergy to SP or to an ACT
2.
An ongoing antibiotic prophylaxis with cotrimoxazole
3.
Current issue requiring hospital admission (including severe malaria as defined by WHO) [17]
4.
Pregnancy at high risk
5.
Women before 16 weeks of pregnancy will not be included because there is insufficient data on the safety of Pyramax® during the first trimester of pregnancy. However, if the WHO changes the recommendations, women in the first trimester could be treated with ACT. In this case, a modification of this protocol will be done accordingly and subjected to ethical review and to competent authorities
Participants will be excluded in case of withdrawal of the informed consent, if they take antimalarial or an antibiotic with antimalarial activity other than those prescribed by the study clinician.
Who will take informed consent?
Pregnant women will be recruited during ANC visits, regardless of their parity and the informed consent will be obtained by the MD of the study team on the field.
Additional consent provisions for collection and use of participant data and biological specimens
It is mentioned in the informed consent form (ICF) that, in addition to the blood collected to perform tests in the field, blood spot will be collected for further examination.
Interventions Explanation for the choice of comparators
This study will compare the IPTp-SP strategy used in routine by the NMCP with the ISTp-US-Py to assess the non-inferiority of the second strategy for the prevention of maternal malaria, maternal anemia, spontaneous abortions or intrauterine death during pregnancy, fetal morbidity (premature birth, low birth weight, small for gestational age), and neonatal mortality at childbirth.
Pyramax® was the perfect drug to use for this procedure as it has been recognized by the NMCP as a drug that can be used during pregnancy in particular although the drug is not used in the overall population.
Intervention description
The ISTp-US-Py group will comprise pregnant women who will be screened monthly from the beginning of the 2nd trimester with us-RDT and who will be treated with Pyramax® if the test is positive.
Pyronaridine-artesunate (Pyramax®, Shin Poong Pharmaceutical Company, South Korea) is a film-coated tablet containing 180 mg of pyronaridine tetraphosphate and 60 mg of artesunate. As part of the EDCTP2 program supported by the European Union (RIA2017MC-2025-Pyrapreg), Shin Poong (Korea) agreed to donate us treatment for this study as well. The IPTp-SP group will be pregnant women who will receive SP as recommended by the National Malaria Control Program (NMCP) at weeks 16, 28, 32, and 36 of their pregnancy. SP (Fansidar®, Roche Laboratories, Switzerland) is a drug used for the treatment of uncomplicated malaria in children and adults and for IPTp. Fansidar is presented as scored tablets containing 500 mg of sulfadoxine and 25 mg of pyrimethamine. The drug continues to be beneficial to both mother and baby, even in areas of SP resistance [18].
Concomitant therapies
Any medication or other therapy taken by the study participant will be recorded.
Criteria for discontinuing or modifying allocated interventions
Changing the distribution of intervention is not possible in the framework of this study. In the event of an allergy or other adverse event (AE) that may impact participants’ health or that of the offspring, the participant will be removed from the study and will no longer receive the intended drug in the arm in which they were allocated.
Strategies to improve adherence to interventions
In order to ensure compliance with the protocol by the study personnel, two monitoring visits are planned throughout the study. The latter will focus on the review of source documents, the distribution of participants by study arm, and the accountability of the study drugs and tests.
Relevant concomitant care permitted or prohibited during the trial
It will be advised to the participants to return to the health center if they feel unwell to receive a concomitant therapy.
Provisions for post-trial care
Coverage of care after the end of the study’s planned follow-up period is scheduled up to 28 days after delivery, though this is not a part of the study but rather for ethical reasons.
Outcomes
Primary outcome
The primary outcome will be the assessment of the malaria status over the pregnancy at every visit for each study participant.
Secondary outcomes
During pregnancy, the following outcomes will be assessed at every visit for each study participant: the anemia status, the incidence of spontaneous abortions, and the intrauterine deaths.
At birth, the outcomes will be assessed as follows in the offspring: the fetal morbidity and the intrauterine death.
Within the 28 days of post-partum, the following outcomes will be assessed in the infant: the early neonatal mortality and the neonatal mortality.
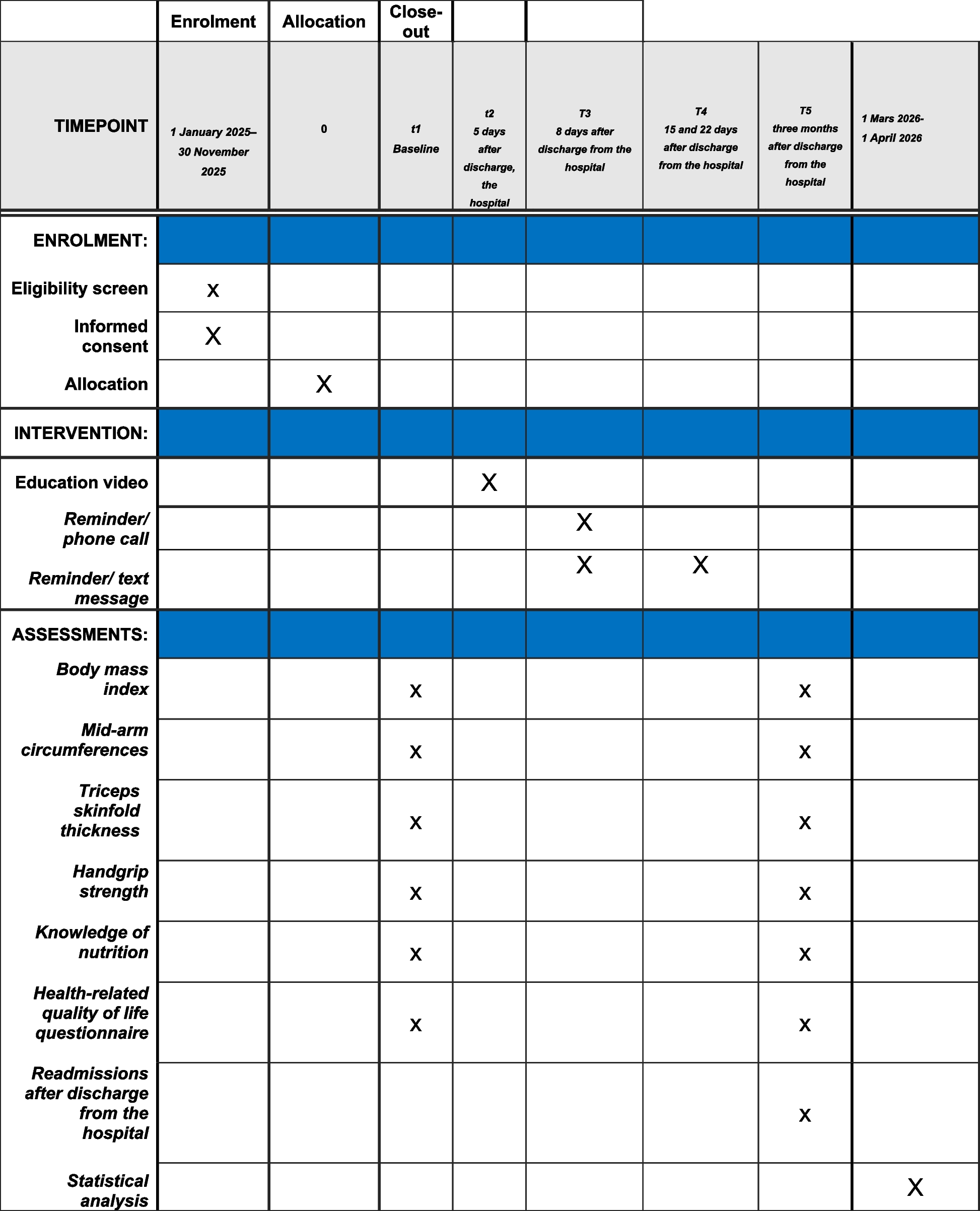
Participant timeline
Pregnant women fulfilling the inclusion/exclusion criteria will be recruited during the ANC visit over an enrollment period of about 20 months. At every scheduled and unscheduled visit, the study team will collect vital signs, blood pressure, weight, data on the medical history since the last visit (including any treatment taken), information on any AE, and current signs and symptoms (if any). At the same time, the team will collect blood samples for malaria diagnosis (us-RDT, thick and thin blood film), Hb, and dried blood spots.
The outcome of pregnancy, birth weight, APGAR score, and maternal Hb will be collected as soon as possible after delivery. A placental blood sample will be collected for the diagnosis of malaria (thin blood film) and dried blood spots. The newborn will be examined for congenital abnormalities and will be reassessed again at day 28 (Table 1).
Table 1 Time and event scheduleSample size
The calculation of the sample size has performed using the Sealed Envelope Ltd. 2012. Power calculator for binary outcome non-inferiority trial. [Online] Available from: https://www.sealedenvelope.com/power/binary-noninferior/.
Referring to the results of a study comparing IPTp-SP and IST (dihydroartemisinin-piperaquine) [11], the number of participants required to detect a difference of 10% with a significance level of 5% and a power of 80% to assess the non-inferiority of IST to IPTp-SP vary as described in Table 2. Variables of interest considered were the malaria status, the anemia status, and the low birth weight rate.
Table 2 Variation of the sample size according to the difference of the outcome of interest
NB: IST with dihydroartemisinin-piperaquine and standard RDT was used as an example to calculate the sample size for the current study as there are no previous studies could be found that include assessment of Pyramax® and us-RDT for use as IST.
Recruitment
Based on the assumption described above, a total of two hundred and twenty pregnant women is the minimal sample size required to answer our research question. However, anticipating on a non-compliance and/or loss-to-follow-up rate of 10%, the adjusted total sample size required will be 242 pregnant women (121 participants per arm).
Assignment of interventions: allocationSequence generation
The study participants will be assigned to the groups using a simple, not stratified randomization of a 1:1 ratio. The randomization list will be generated by an independent data technician using Excel Microsoft prior to the inclusion.
Concealment mechanism
The randomization numbers will be stored in sealed individual envelopes that will be opened in front of the participants.
Implementation
The randomization list will be generated by a data manager non-involved in the study.
Assignment of interventions: blindingWho will be blinded
All the participants will be aware of the drug regimen they will be assigned to. But the statistician will be blinded for the analysis to avoid any bias.
Procedure for unblinding if needed
Not applicable. The study team and the participants themselves will all be aware of the arms where participants will be included.
Data collection and managementPlans for assessment and collection of outcomes
Pregnant women fulfilling the inclusion/exclusion criteria will be recruited during ANC in the study site over a period of about 20 months. Data will be entered by the MDs in paper CRF designed by the study site in accordance to variables to collect over the participants’ visits during the study. Later, data managers will be in charge to enter the data collected in papers in the electronic database. To ensure the accuracy of the process, source data verification of every entry will be performed by the study supervisors before the validation of the database.
At every scheduled and unscheduled visit, the study team will collect vital signs, blood pressure, weight, data on the medical history since the last visit (including any treatment taken), information on any AE, and current signs and symptoms (if any). At the same time, the team will collect blood samples for malaria diagnosis (us-RDT, thick and thin blood film), Hb, and dried blood spots for PCR.
The outcome of pregnancy, birth weight, APGAR score, and maternal Hb will be collected as soon as possible after delivery. Dried blood spots will be collected on placental blood. The newborn will be examined for congenital abnormalities. Both the mother and the newborn will be reassessed at day 28.
Demographic data and medical history
Demographic data and a general history of past and/or present illnesses, the medical history since the last visit (including any treatment taken), information on any AE, and current signs and symptoms (if any) will be recorded.
Physical and clinical examination
Vital signs (body temperature, pulse rate), blood pressure, and weight will be measured.
An obstetrical examination will be performed and the fetal viability will be recorded.
Bloodspots of 50 μl will be collected on filter paper (Whatman grade 3) at each visit and subsequently every time a blood slide is done. Samples will be used to determine sub-microscopic parasitemia.
In addition to an us-RDT detecting circulating P. falciparum antigens, thick and thin blood films will be prepared, dried, and stained with Giemsa according to standard operating procedures (SOP).
If positive, the parasite density (PD) will be calculated by counting the number of P. falciparum parasites per 200 leukocytes based on the actual number of WBC/μl. In case this information is missing, the PD will be estimated assuming WBC of 8000/μl. The PD per microliter will be calculated using the following formula:
$$\mathrm/\mathrm\mathrm(\mathrm P/\mathrm\mathrm)=\frac\;\mathrm\;\mathrm p\mathrm a\mathrm r\mathrm a\mathrm s\mathrm i\mathrm t\mathrm e\mathrm s\;\mathrm c\mathrm o\mathrm u\mathrm n\mathrm t\mathrm e\mathrm d\times8,000}$$
The thin smear will be examined for species determination.
Blood smears will be independently read by 2 qualified microscopists and the mean of the PD will be used in the study. Further details are described and explained in the SOP (SOP: quality control (QC) guidelines for the laboratories participating in the study).
Plans to promote participant retention and complete follow-up
Participant’s visits are scheduled on monthly basis. In case of a missing visit, the site clinicians will be in charge to phone the participants to enquire for the reason of the missing visit and encourage them to come to the study site. In case participants cannot be reached by phone, a community health worker will be visiting the households for enquiring and encourage participants to come to the study site. In case participants are not reachable, they will be considered as lost to follow-up after 3 missing visits and all attempts to reach out to them will be documented.
In case of loss to follow-up, all the data collected during the woman participation in the study will be used for the analysis. However, in case of consent withdrawal, the participant data will not be used for the analysis.
Data management Data management and storage
Data management will be handled by the University of Kinshasa which has the required expertise in data management of clinical trials and epidemiological studies.
Data entry and review will be performed following the Data Entry Guidelines and the Data Management plan. Besides this central management, two monitoring visits are planned to check the information entered into the electronic CRF against the source documents available on site. Any modification done onto the electronic CRF will be automatically registered. The final database will be obtained after the resolution of all queries and will be locked for statistical analysis to be carried out according to a pre-established data analysis plan that will be developed by a statistician and submitted for comments and advice to the Trial Steering Committee (TSC).
Every patient will have a personal source document file. Data will be collected onto the source document and entered later in an electronic CRF. Data will be entered on a daily basis on the database.
Study documents will be archived for 10 years and granted to the sponsor for trial-related monitoring, audits, DSMB, and Ethical Committee review and regulatory inspections when applicable.
Confidentiality
Collected personal information will be restricted to meet the objectives of the study. Prior to the study start, all study staff will sign a confidentiality agreement form. Every participant will be assigned a unique study identification code and no name or personal information will appear in the database or will ever be published. All personal information mentioned in the signed informed consent forms and other documents will be kept under lock and will only be available to the project coordinator and data managers. Information stored in the database will be protected by unique usernames and passwords, which will only be available to minimum appropriate authorized personnel.
Plans for collection, laboratory evaluation, and storage of biological specimens for genetic or molecular analysis in this trial/future use
For this study, the laboratory analysis carried out will be the Hb dosage using a Hemocue, a diversified evaluation of malaria by TDR, US-TDR, thick and thin smear, and PCR.
Hb assay, TDR, US-TDR, and blood smear will be done on-site. Blood will be taken from a dried blood spot for further PCR analysis. The dried blood spot will be stored in individual zippers, each coded for the study, stored in a dry place and containing silica gel to prevent spoilage.




留言 (0)