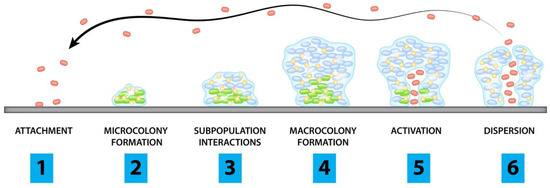
2.1.1. Degradation of Viral ProteinIn this section, PROTACs that target viral proteins have been enlisted. PROTAC-based protein degraders are highly explored in this section as compared to other strategies of protein degradation. Many viral proteins have been targeted for protein degradation (
Table 2). For instance, Montrose et al. developed a peptide-based PROTAC molecule that targets X-protein, which is an essential protein required for the replication of the hepatitis B virus (HBV). It was also found that the presence of X-protein could also induce hepatocellular Carcinoma (HCC). This PROTAC molecule consists of an ODD degrons (oxygen-dependent degradation) domain, an oligomerization domain, and a cell-penetrating peptide. The ODD degrons domain binds to Von Hippel–Lindau (VHL) E3 ligase, the oligomerization domain interacts with the X-protein, and octa-arginine is used as a CPP for to ease cellular entry. In vitro studies verified the ability of peptide-based PROTACs to efficiently degrade X-protein [
48]. In another study, instead of the peptide as a ligand for POI, the authors used telaprevir, an anti-viral peptidomimetic protease inhibitor. They developed three different molecules (DGY-03-081 (
2), DGY-04-035 (
3), and DGY-08-097 (
4)) that target the NS3/4A protease of the hepatitis C virus (
Figure 2). Lenalidomide, pomalidomide and novel tricyclic imide moiety were used as the ligands for CRBN E3 ligase. The function of NS3/4A serine protease is to cleave viral polyprotein, which acts as an essential step in viral replication [
49]. Thus, degradation of NS3/4A protease via PROTAC will inhibit virion formation and multiplication. These compounds were evaluated in Hep C virus-infected HEK293T cells. Interestingly, all three degraders exhibited anti-viral activity and did not show cytotoxicity to the uninfected cells. Compound DGY-08-097 (
4) had the highest degradation ability and the least DC50 value (50 nM at 4 h). One of the reasons for the increased affinity might be due to the tricyclic imide moiety in the DGY-08-097 (
4) that showed increased affinity towards CRBN E3 ligase [
50].Similarly, the endonuclease polymerase subunit (PA) of influenza virus A was the POI for developing a novel PROTAC molecule. Asperphenalenone E (APL-16-5) (
5) was derived from an endophytic fungus Aspergillus, which was used as a ligand for PA. APL-16-5 (
5) induces degradation of the viral polymerase subunit (PA) by ubiquitin–proteasome machinery, as it can bind to both the E3 ligase enzyme (TRIM25) and PA. The endonuclease polymerase enzyme is essential in the polymerization of DNA during DNA replication. Derivatives of asperphenalenone, APL-16-1 (
6), and APL-16-2 (
7) were also synthesized, and the results were compared with the known anti-viral drug ribavirin. HEK293T, A549, and MDCK cells were cultured and infected with influenza virus A WSN/33 for in vitro analysis. The cytotoxicity of APL-16-5 (
5) and APL-16-1
(6) against the Influenza virus was in micromolar concentration (EC50) 0.28 to 0.36 μM. Proteosome-mediated degradation of PA with APL-16-5 (
5) exhibited a marked decrease in viral RNA components. Later, APL-16-5 (
5) was evaluated against influenza virus B, hepatitis C, and Zika viruses. The results from the study confirmed that APL-16-5 is a selective inhibitor for influenza viruses. Dose-dependent studies were conducted to determine the interaction of PA with TRIM25 and concluded that compound
5 induces the destabilization of PA by ubiquitination, and thereby it degrades the PA [
51].Li et al. designed a pentacyclic triterpenoid group (PTG) containing PROTAC molecule for targeting hemagglutinin (HA) of the influenza virus. Pentacyclic triterpenoids are secondary metabolites present in various medicinal plants, and they possess significant anti-viral activity. Oleanolic acid (OA) and its derivatives are compounds that were selected as the warhead for the PROTAC molecule. OA exhibited anti-viral action against the influenza A/WSN/33 virus, and it had a moderate binding affinity with HA; thus, it became an ideal molecule for PROTAC technology. Two sets of PROTAC molecules (
8–
10) and (
11–
16) were designed and studied employing different E3 ligases, such as CRBN and VHL ligands, respectively. HEK293T cells were transfected using HA plasmids, and the level of HA degradation was studied using the synthesized PROTAC molecules. A cell viability assay, immunofluorescence microscopy assay, immunoprecipitation assay, hemagglutination inhibition assay, etc., were performed to evaluate the molecules. Compound
13 (DC50 = 1.44 μM) exhibited the maximum HA depletion as compared to other compounds. This was also validated by molecular docking analysis by Schrodinger Suite. Furthermore, it was concluded from these assays that the VHL ligand containing PROTACs showed better HA degradation [
52].In another independent study conducted by Xu et al., oseltamivir is an approved drug for influenza that targets influenza neuraminidase (NA). Neuraminidase is an essential enzyme for viral replication. They have used oseltamivir-based compounds for targeting neuraminidase and linked them with a discrete variety of E3 ligase ligands such as VHL or CRBN. The amino or carboxylate group of oseltamivir was modified to improve its anti-viral activity. A wide variety of linker combinations like rigid as well as flexible groups like PEG, pyridyl, triazole, and piperazinyl were also involved. A set of PROTAC combinations (
17–
38) were designed, and from these, N-substituted oseltamivir showed increased potency than the carboxylate-substituted compound. According to the in vitro studies, compound
27 showed the best anti-viral activity having an EC50 of 0.33 µM, which was almost similar to the reference drug oseltamivir phosphate (EC50 = 0.36 µM). Furthermore, interestingly, all the synthesized compounds do not show cytotoxicity towards the normal cells with a concentration up to CC50 > 50 µM. Docking studies indicated that these ternary complexes showed great hydrogen bonding and hydrophobic interactions between neuraminidase and E3 ligase [
53]. From these above studies, it could be concluded that there are various strategies being evolved to target viral proteins and inhibit their replication. Other interesting subcategories of PROTAC are ribonuclease-targeting chimera (RIBOTAC) and nucleic acid-hydrolysis-targeting chimera (NATAC). Both strategies were used to develop novel degraders. In RIBOTAC, RNase is the degrader system, and it degrades viral RNA, while NATAC uses oligonucleotide sequences to identify the POI, and further, they could be degraded by RNase L (specific for ss-RNA). Haniff et al. developed a RIBOTAC degrader that targets the RNA genome of the SARS-CoV-2 virus (
Figure 3). RIBOTAC has two major constituents- a small molecule known as C5 (
39) and an RNA attenuator hairpin (AH). This RNA attenuator hairpin binds to the RNA genome, and C5 (
39) recruits endonucleases present in the cell and initiates the degradation of the viral genome (
Figure 4) [
54]. This strategy might provide solutions for various viral infections, and the only challenge is identifying and optimizing the appropriate attenuator sequence, which could bind toward a target of interest. NATAC is another promising approach to PROTAC technology (
Figure 5). NATAC explores the function of RNase L; RNase L is an innate RNA degrading enzyme that targets UN^N sites of the viral or single-stranded RNA. Tang and his group designed a NATAC molecule having a 5’ phosphorylated 2’-5’ polyA sequence that attracts RNase L, and another end had an antisense oligonucleotide strand targeting the spike protein of SARS-CoV-2 (Compound
40) (
Figure 4). The knockdown efficiency of spike proteins by NATAC molecules was evaluated, and it was found that NATACs could significantly reduce the RNA sequence of the spike protein. Interestingly, it was also found that RNase L could also increase the mRNA level of IFN-β and IL-6 in the host cells, thereby increasing the production of interferon and further enhancing the anti-viral response in the host cell [
55]. 2.1.2. Degradation of Human Host ProteinIn this section, PROTACs that target human host cell proteins, which are involved with viral proteins to enhance their virulence, are listed (
Table 3). For instance, human cytomegalovirus (HCMV) is one of the major pathogens that cause the herpes disease. It was found that HCMV protein kinase pUL97, a viral cyclin-dependent kinase (vCDK), plays a crucial role in the generation of nuclear capsid and viral replication. During HMCV infection, viral proteins upregulate the expression of various CDK-cyclin complexes that initiate pseudomitosis, which is favorable for viral replication. Thus, CDK inhibitors targeting CDKs could be a solution for HCMV infection. In this study, THAL-SNS032, which is a protein kinase inhibitor, was used to target CDK9, and thalidomide was the E3 ligase targeting ligand. In vitro cytotoxicity studies showed that the THAL-SNS032 (EC50 0.025 ± 0.001 µM) was nearly fourfold more efficient than the nonPROTAC parent compound SNS032 (EC50 = 0.105 ± 0.004 µM) [
56]. Thus, it could be concluded that these PROTAC molecules could be a possible candidate for treating herpes disease.The research community was doomed due to the pandemic SARS-CoV-2 and researchers across the globe were trying to provide solutions for COVID-19. This led to the discovery of a repurposing drug, i.e., indomethacin (IMN) (Compound
41), which is an anti-inflammatory drug and was found to have anti-viral activity against the Coronaviridae family (
Figure 6). Desantis et al. designed four PROTAC molecules (
42–
45) using indomethacin for targeting human prostaglandin E synthase type 2 (PGE-2). PGE-2 interacts with the NSP7 protein of SARS-CoV-2. NSF7 proteins are essential for SARS-CoV-2 replication. However, the exact mode of action of indomethacin is unknown. Furthermore, it is confirmed that degradation of PGE-2 inhibits repression of viral protein synthesis by ds-RNA-dependent protein kinase R (PKR)-mediated pathway. Since this PROTAC degrades human cellular protein, it could be classified as PROTACs targeting host protein for degradation. These four synthesized PROTAC compounds (
43) and (
45) showed the highest activity, with EC50 values of 18.1 and 21.5 μM, which was nearly five times more effective than indomethacin (EC50 = 94.4 μM). Molecular docking studies also described that using 6 methylene units in (
43) and a piperazine group in (
45) as a linker seems to be perfect for the molecule’s binding interaction with the VHL ligand. Moreover, compounds (
43) and (
45) exhibited anti-viral activity against β-coronavirus (i.e., HCoV-OC43), with EC50 values of 4.7 and 2.5 µM, respectively. These compounds were specific in that they did not show cytotoxicity against MRC-5 cells (normal uninfected cells), indicating that these PROTACs are able to specifically target infected cells. PROTACs showed effective broad-spectrum action in inhibiting two different SARS-CoV-2 strains, i.e., β-coronavirus HCoV and the α-coronavirus HCoV-229E. Due to the broad-spectrum nature of the PROTACs, these molecules could be a potential target for COVID-19 disease [
57]. Y et al. developed a novel PROTAC targeting HUWE1 E3 ligase and ORF3 MERS-CoV accessory protein. ORF3 restricts apoptosis in the host cell, which could be beneficial for the virus to replicate. Hence, degradation of the ORF3 protein will induce apoptosis in the host cell, thereby restricting the spread of MERS-CoV infection. HUWE1 E3 ligase was specific towards the degradation of ORF3 since PROTACs targeting other E3 ligases such as UBR5, TRIM33, Cullin5, Cullin3, and UBR4 had no effect on the stability of ORF3. Thus, it indicates that HUWE1 E3 ligase could be a better E3 ligase for targeting ORF3 of MERS-CoV. Subsequently, MERS-CoV belongs to the same family as SARS-CoV-2; studies on SARS-CoV-2 infected cells could be another potential area of research for targeting COVID-19 [
58].



留言 (0)