1. IntroductionThe retina expends excessive energy for the formation of visual perception. It is very sensitive to oxidative stress. At the same time, the retina serves as a powerful generator of reactive oxygen species (ROS) implicated in several major retinal diseases, including age-related macular degeneration (AMD), a leading cause of vision loss [
1,
2]. Although the etiology and mechanism of AMD induction remain unclear, oxidative stress-related injury to the retinal pigment epithelium (RPE) is recognized as an early event in in AMD-like pathology [
3,
4]. Appropriate levels of intracellular ROS including mitochondrial ROS (mtROS) play important physiological roles as modulators of cellular signaling pathways. However, excessive accumulation of ROS by persistent oxidative stress can lead to cellular injury and death and contribute to the initiation of pathological damage to various organs, including eyes [
5,
6]. In addition, apoptosis and autophagy of RPE cells due to excessive ROS production are accompanied by mitochondrial and DNA damage, ultimately contributing to retina dysfunction [
7,
8]. Furthermore, since mitochondrial damage in RPE degeneration can induce a cellular defense mechanism known as mitophagy, mitophagy could be a putative therapeutic target in retinal degenerative diseases such as AMD [
9,
10]. Therefore, the level of ROS must be tightly controlled to protect normal functions of eyes.Natural resources have long received great attention as sources of drug development. Among them, phenolic compounds derived from natural products having excellent antioxidant activity have attracted attention. Their antioxidant activities mainly involve scavenging of ROS and activation of intracellular antioxidant signaling pathways [
5,
11,
12]. Phloroglucinol, a polyphenol trihydroxybenzene with an aromatic phenyl ring and three hydroxyl groups, is a naturally occurring secondary metabolite present in a variety of organisms including plants, algae, and bacteria [
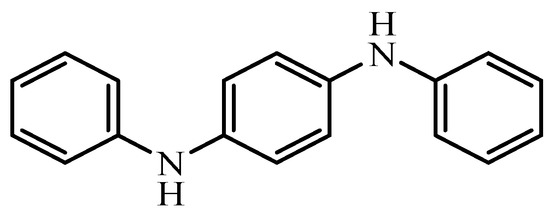
13,
14]. This phenolic compound is known to have various pharmacological potentials such as antibacterial, anticonvulsant, anti-allergic, antithrombotic, anti-inflammatory, and cancer chemopreventive activities [
15,
16]. Recently, the antioxidant potential of phloroglucinol has been validated in several in vitro and in vivo models. For example, Drygalski et al. [
17] have reported that phloroglucinol can strengthen antioxidant defense and ameliorate hepatic steatosis and inflammatory response by reducing oxidative/nitrogen damage to cellular macromolecules. In addition, it has been confirmed that phloroglucinol can block oxidative damage caused by hydrogen peroxide (H2O2) treatment and γ-ray irradiation by regulating activities of antioxidant and detoxifying enzymes in the retinal epithelium, hippocampal nerve, renal epithelial cells, and lung fibroblasts [
18,
19,
20]. Moreover, phloroglucinol as an ROS scavenger can modulate synaptic plasticity to attenuate pathological phenomena of neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease [
21,
22]. Our previous study has shown that phloroglucinol can inhibit DNA damage and apoptosis in H2O2-exposed HaCaT human keratinocytes [
23]. Similar results have been confirmed in ultraviolet (UV) B-irradiated keratinocytes and all-trans-retinal-exposed primary rat RPE and mouse photoreceptor cells [
19,
24]. Recently, Kuo et al. [
25] have reported that phloroglucinol can block oxidative cytotoxicity induced by potassium bromate, an AMD inducer, in human RPE ARPE-19 cells by inhibiting ROS production. These results suggest that phloroglucinol could play an antioxidant role in ARPE-19 cells as suggested by Moine et al. [
26]. Nevertheless, studies on the protective role of phloroglucinol against cellular damage induced by oxidative stress in RPE cells are lacking. Therefore, the purpose of the current study was to investigate effects of phloroglucinol on oxidative stress-induced mitochondrial and DNA damage and induction of apoptosis and autophagy in RPE cells. For this purpose, a human RPE-derived ARPE-19 cell model treated with H2O2 to mimic oxidative stress was used. 2. Materials and Methods 2.1. Cell Culture and TreatmentARPE-19 cells (CRL-2302) were purchased from the American Type Culture Collection (Manassas, VA, USA) and routinely cultured in Dulbecco’s Modified Eagle Medium/F-12 supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin (WELGENE Inc., Gyeongsan, Republic of Korea) as described previously [
27]. To investigate beneficial effects of phloroglucinol on oxidative damage, cells were cultured in media containing desired concentrations of phloroglucinol and H2O2 (Thermo Fisher Scientific, Inc., Waltham, MA, USA) for 24 h or pretreated with phloroglucinol, N-acetyl-L-cysteine (NAC), Mito-TEMPO, and/or 3-methyladenine (3-MA, Sigma-Aldrich Co., St. Louis, MO, USA) for 1 h prior to treatment with H2O2 for 24 h. To investigate the blocking effect of phloroglucinol on the generation of ROS induced by H2O2, cells were pretreated with phloroglucinol, NAC, and Mito-TEMPO for 1 h and then treated with H2O2 for 1 h. To acquire fluorescence images of ROS generation, γH2AX expression, and autophagic vacuoles, cells cultured on coverslips were stimulated with H2O2 in the presence or absence of phloroglucinol, NAC, and/or Mito-TEMPO. After treatment, cells were washed with phosphate-buffered saline and subjected to fluorescence staining. 2.2. Cell Viability AssayTo investigate viability of ARPE-19 cells cultured under various conditions, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetra-zolium bromide (MTT) assay was performed. In brief, after the necessary experimental treatment, cells were incubated with MTT solution (Thermo Fisher Scientific, Inc.) for 3 h. Formed insoluble formazan products were then dissolved in dimethyl sulfoxide (DMSO, Thermo Fisher Scientific, Inc.) and the absorbance was read at 570 nm using an enzyme-linked immunosorbent assay (ELISA) microplate reader (Molecular Device Co., Sunnyvale, CA, USA) according to a previously described method [
28]. Cell viability was expressed as a percentage of untreated control cells. 2.3. Cytotoxicity Assay
To assess cytotoxicity, lactate dehydrogenase (LDH) release was detected using an LDH Cytotoxicity Assay Kit (Thermo Fisher Scientific, Inc.) according to the manufacturer’s instructions. In brief, culture medium obtained from conditions treated with H2O2 in the presence or absence of phloroglucinol was transferred to a 96-well plate and the amount of released LDH was measured at 490 nm with an ELISA microplate reader.
2.4. Quantitative Assessment of ApoptosisAnnexin V-Fluorescein Isothiocyanate (FITC) Apoptosis Detection Kit was purchased from Abcam Inc. (Cambridge, UK) and used for quantitative evaluation of apoptosis-induced cells upon treatment with phloroglucinol and/or H2O2. After treatment, collected cells were suspended in annexin binding buffer containing annexin V- FITC and propidium Iodide (PI) following the manufacturer’s instructions. The fluorescence of 10,000 events was then acquired using a flow cytometer (Becton Dickinson, San Jose, CA, USA). Annexin V-positive cells were considered as apoptosis-induced cells as described previously [
27]. 2.5. DNA Fragmentation AssayTo observe fragmented DNA, an apoptosis marker, cell pellet was suspended in a lysis solution as described previously [
29]. The supernatant was then incubated with RNase A and proteinase K (Sigma-Aldrich Co.). DNA was precipitated with isopropyl alcohol (Sigma-Aldrich Co.). The extracted DNA was fractionated using 1.0% agarose gel and then stained with ethidium bromide (EtBr, Thermo Fisher Scientific, Inc.) to visualize DNA fragmentation pattern, a characteristic of apoptosis, under UV light. 2.6. Protein Isolation and Western BlottingTotal protein to be used for Western blot analysis was extracted as described previously [
30]. Cytoplasmic and mitochondrial proteins were isolated using a Mitochondrial Fractionation Kit (Thermo Fisher Scientific, Inc.) following the manufacturer’s instructions. In brief, proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to Immobilon®-P PVDF membranes (Merck Millipore, Bedford, MA, USA). These membranes were then incubated with specific primary antibodies overnight followed by reaction with horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) at room temperature for 1 h. Immune complexes were visualized with enhanced chemiluminescence reagent (Thermo Fisher Scientific, Inc.) according to the manufacturer’s instruction [
31]. Densitometric analysis of the data was performed using the ImageJ® software (v1.48, NIH, Bethesda, MD, USA). Primary antibodies were obtained from Santa Cruz Biotechnology, Inc. and Cell Signaling Technology (Beverly, MA, USA). All antibodies used in this study are listed in
Table 1. 2.7. Caspase-3 Activity AssayCaspase 3 activity was quantified using a Caspase-3 Colorimetric Assay Kit (Abcam, Inc.). In brief, aliquots of cytosolic extracts were mixed with a fluorescent substrate of caspase-3, acetyl-Asp-Glu-Val-Asp-chromophore-p-nitroanilide (Ac-DVAD-pNa), in the buffer provided in the kit according to the manufacturer’s instructions. Enzyme-catalyzed release of pNa was monitored at 405 nm using an ELISA microplate reader. The activity of caspase-3 was presented relative to the control [
32]. 2.8. Assessment of ROS Generation
Levels of intracellular ROS and mtROS production were detected using fluorescent probes 2′,7′-dichlorofluorescein diacetate (DCF-DA) and MitoSOX (Sigma-Aldrich Co.), respectively. Following exposure to H2O2 with or without phloroglucinol, NAC, and/or Mito-TEMPO, cells were reacted with DCF-DA and MitoSOX to assess levels of intracellular and mitochondrial peroxides, respectively, using flow cytometry. In parallel, fluorescence images of DCF-DA- and MitoSOX-stained cells cultured on coverslips were observed under a fluorescence microscope (Carl Zeiss, Oberkochen, Germany) at Core-Facility Center for Tissue Regeneration, Dong-eui University (Busan, Republic of Korea).
2.9. Comet Assay
The inhibitory effect of phloroglucinol on H2O2-induced DNA damage after appropriate treatment was determined using comet assay (single cell gel electrophoresis). Briefly, collected cells were suspended in 1% low melting point agarose and then spread on comet slides according the manufacturer’s protocol in of a commercially available Comet Assay Kit (Trevigen, Inc., Gaithersburg, MD, USA). After DNA denaturation, electrophoresis was performed and slides were stained with an asymmetric cyanine dye. Resulting images were acquired under a fluorescence microscope.
2.10. γH2AX Immunofluorescence Assay
Immunofluorescence assay was applied to analyze the expression of phosphorylated histone H2AX (p-γH2AX) in cells treated with or without phloroglucinol or NAC before adding H2O2. Following treatment, cells were fixed with formaldehyde, permeabilized with Triton X-100 solutions (Thermo Fisher Scientific, Inc.), and then blocked with bovine serum albumin solution (Sigma-Aldrich Co.). Thereafter, cells were probed with an anti-p-γH2AX antibody (Cell Signaling Technology, Inc.) and then reacted with Alexa Fluor 555-conjugated secondary antibody (Thermo Fisher Scientific, Inc.). For nuclear counterstaining, cells were immersed in a 4’,6-diamidino-2-phenylindol (DAPI) solution (Sigma-Aldrich Co.). Then, p-γH2AX and DAPI fluorescence images were captured using a fluorescence microscope.
2.11. Measurement of 8-Hydroxy-2′-Deoxyguanosine (8-OHdG)
To measure 8-OHdG, a deoxyriboside form of 8-oxoGuanine, an OxiSelect Oxidative DNA Damage ELISA Kit (Cell Biolabs, San Diego, CA, USA) was used. Briefly, DNA was extracted from cells cultured under the same conditions as described above. Subsequently, the DNA of each isolated sample was digested with DNase I (Sigma-Aldrich Co.). The absorbance of the ELISA reaction was then measured at 450 nm following the protocol presented in the kit.
2.12. Mitochondrial Membrane Potential (MMP) Measurement
MMP level was monitored by staining with 5,5′,6,6′-tetrachloro-1,1′3,3′-tetraethyl- imidacarbocyanune iodide (JC-1), a fluorescent carbocyanine probe. For this assay, cells treated with H2O2 in the presence or absence of phloroglucinol or Mito-TEMPO were stained with JC-1 solution (Thermo Fisher Scientific, Inc.). The percentage of JC-1 monomer was analyzed with a flow cytometer to indicate cells that lost MMP.
2.13. Autophagy Detection
Formation of autophagosomes was assessed using a CYTO-ID® Autophagy Detection Kit purchased from Enzo Life Sciences, Inc. (Farmingdale, NY, USA). First, cells were collected for quantitative analysis of autophagy induction. Cyto-ID staining procedure was then performed according to the manufacturer’s instructions. In brief, cells cultured under various conditions were washed with assay buffer included in the kit and fixed with paraformaldehyde. Fluorescently labeled cells were then analyzed by flow cytometry. Next, cells were subjected to DAPI staining after CYTO-ID staining to monitor localizations of autophagosomes and nuclei. To monitor localizations of autophagosomes and nuclei, cells were further subjected to DAPI staining after CYTO-ID staining. The autophagic signal (green) and the nuclear signal (blue) were collected under a fluorescence microscope.
2.14. Statistical Analysis
All statistical analyses were performed using GraphPad Prism (Graphpad Inc., San Diego, CA, USA). Statistical differences were determined by one-way analysis of variance with Tukey’s test. Statistical significance was considered when p-value was less than 0.05. All data are expressed as mean ± standard deviation (SD) (* p < 0.05, ** p < 0.01 and *** p < 0.001 vs. unstimulated control; ##p < 0.01 and ###p < 0.001 vs. H2O2 alone treatment; &p < 0.05 and &&&p < 0.001 vs. phloroglucinol + Mito-TEMPO group).
4. Discussion
In the current study, we induced oxidative stress using H2O2 to examine whether phloroglucinol could protect human RPE ARPE-19 cells from oxidative injury. We found that H2O2 induced apoptosis, accompanied by mitochondrial dysfunction, DNA damage, and autophagy through an increase in ROS generation. However, phloroglucinol was able to block H2O2-induced cellular damage and scavenge ROS.
Induction of cytotoxicity including DNA damage and cell death by oxidative stimulation is mostly accompanied by mitochondrial dysfunction associated with ROS generation [
33,
34]. In healthy retinal cells, ROS levels remain low as a result of normal cellular metabolism. However, accumulation of ROS caused by oxidative stress can act as an initiator in the pathogenesis of degenerative diseases of the retina [
4,
35]. In this study, inhibition of cell survival, induction of cytotoxicity, and generation of ROS by H2O2 in ARPE-19 cells were significantly suppressed by pretreatment with phloroglucinol or NAC, a free-radical scavenger used as a positive control. These results showed the possibility that phloroglucinol could block ROS generation caused by oxidative stress. Many previous studies have shown that DNA damage and apoptosis can be induced in RPE cells exposed to oxidative stimuli [
35,
36]. This finding was also confirmed in H2O2-treated ARPE-19 cells. We first performed a comet assay, a widely used method to detect DNA strand breaks in eukaryotic cells [
37], to evaluate whether pretreatment of phloroglucinol could inhibit H2O2-induced DNA damage. We found that phloroglucinol effectively inhibited the comet tail moment (DNA migration) observed in cells treated with H2O2. In addition, the expression of p-γH2AX, a biomarker of DNA double-strand break [
38], and the amount of 8-OHdG, an indicator of oxidative stress-mediated DNA damage [
39], were increased by H2O2 treatment. However, these changes were all canceled by treatment with phloroglucinol. The blocking effect of phloroglucinol on these three indicators was similarly observed in cells pretreated with NAC. Our results well support results shown in H2O2-treated human keratinocytes and UVB-irradiated mouse skin [
18,
23,
24]. These results suggest that the ROS scavenging ability of phloroglucinol might contribute to the reduction in H2O2-induced DNA damage in RPE cells.Apoptosis is usually divided into extrinsic and intrinsic pathways. Overload of ROS by oxidative stress can depolarize the mitochondrial membrane, which contributes to the activation of mitochondria-mediated intrinsic apoptosis pathway [
40,
41], resulting in the collapse of MMP indicative of dysfunctional mitochondria and leading to cytosolic release of cytochrome c. Released cytochrome c can activate the caspase cascade required for the intrinsic apoptosis pathway, causing degradation of caspase-dependent proteins such as PARP, thereby terminating apoptosis [
40,
42,
43]. As reported in previous studies [
23,
44,
45], the reduction in MMP and cytoplasmic release of cytochrome c are major events during mitochondria-mediated apoptosis. These events were increased in H2O2-treated ARPE-19 cells in the present study. However, there changes were markedly blocked by phloroglucinol. Furthermore, expression of Bcl-2 family proteins, activation of caspase-3, and cleavage of PARP by H2O2 were maintained at control levels after phloroglucinol pretreatment, in good agreement with our previous study using human keratinocytes [
23]. Accumulated prior studies have shown that the intrinsic pathway is critically controlled by Bcl-2 family members. Among them, anti-apoptotic proteins including Bcl-2 are essential to maintain stability of the mitochondrial membrane barrier, whereas anti-apoptotic proteins such as Bax are key executors of mitochondrial poration, thereby enhancing mitochondrial membrane permeability and releasing mitochondrial cytochrome c [
8,
40]. These findings well support our finding that phloroglucinol can prevent apoptosis by suppressing the intrinsic apoptotic pathway. Taken together, our findings indicate that the antioxidant activity of phloroglucinol is responsible for H2O2-induced blockade of apoptosis in ARPE-19 cells.Although the primary targets of intracellular ROS are mitochondria, mitochondria are also major sources of ROS. Increased ROS in turn can inhibit mitochondrial efficiency, which can lead to more ROS production in mitochondria by a self-destructive vicious cycle [
46,
47]. Therefore, we evaluated whether ROS generated by H2O2 was derived from mitochondria by applying MitoSOX-red, a mitochondrial superoxide indicator, and Mito-TEMPO, a specific antioxidant for mtROS based on previous studies showing that the generation of ROS induced by H2O2 in ARPE-19 cells occurs in mitochondria [
48,
49]. MMP lost by H2O2 was also significantly abolished by treatment with phloroglucinol or Mito-TEMPO alone. However, in cells pretreated with both phloroglucinol and Mito-TEMPO, MMP was almost completely restored to untreated control levels. Moreover, H2O2-induced cytosolic release of cytochrome c and expression of mitophagy markers such as PINK1 and PARKIN were not observed in cells pretreated with phloroglucinol, which might be due to blockade of mtROS production by phloroglucinol. During mitophagy, a type of autophagy unique to mitochondria, PINK1 recruits PARKIN for autophagosome formation, which in turn initiates the removal of damaged mitochondria via autophagy and proteasome mechanisms [
50,
51]. In AMD-like pathology associated with RPE injury, accumulation of mitochondrial damage and reduction in biogenesis are closely related to the induction of mitophagy, a phenomenon that appears prominently as aging progresses [
4,
10]. In particular, an aged retina is characterized by increased ROS accumulation, impaired autophagy, and mitochondrial damage associated with the pathogenesis of AMD. Rohrer et al. [
52] have demonstrated that RPE cells isolated from eyes of elderly donors are more sensitive to oxidative stress and that a further decrease in mitochondrial metabolism might be associated with increased mitophagy. In addition, Kim et al. [
9] have recently shown that mitochondrial dysfunction in H2O2-injured rat retina and RPE cells is responsible for the induction of mitophagy. As in other cells, oxidative stress-induced mitophagy in RPE cells occurs through the PINK1-PARKIN signaling pathway, a process that clears damaged mitochondria through autophagy [
53,
54]. Therefore, our results suggest that suppression of mtROS production and preservation of mitochondrial function by phloroglucinol in ARPE-19 cells exposed to H2O2 are mediated by blockade of mtROS production.Recently, the importance of autophagy in AMD pathology has been steadily rising. It has been shown that mtROS-mediated autophagy induced by oxidative stress may contribute to retinal damage [
55,
56]. Autophagy is a critical catabolic process for adapting to metabolic stress and maintaining homeostasis by removing damaged intracellular organelles (including mitochondria) and proteins through formation of autophagosomes. This process is involved in the promotion and inhibition of apoptosis depending on stimulators that induce autophagy, the type of cell, and the environment surrounding the cell [
50,
51]. One of the features of retinal aging is the accumulation of autophagy proteins associated with mitochondrial damage [
57,
58]. In this respect, pharmacological manipulation of autophagic activity could be a therapeutic target for retinal damage-related disorders. Although autophagy in RPE cells exposed to oxidative stress, particularly H2O2, is known to contribute to apoptosis induction [
49], RPE cells might also be protected from oxidative stress and apoptosis through promotion of autophagy [
59]. In this study, H2O2-induced autophagy in ARPE-19 cells was blocked by 3-MA, an autophagosome blocker, suggesting that H2O2-mediated autophagy might contribute to apoptosis induction. Phloroglucinol also conferred a protection against H2O2-induced autophagy, similar to 3-MA. In addition, as is commonly observed during autophagy, in H2O2-treated cells, the conversion of LC3-I to LC3-II was increased and p62 was down-regulated while Beclin-1 was up-regulated. However, H2O2 stimulation in the presence of phloroglucinol failed to induce an increase in LC3-II/LC3-I value or Beclin-1 expression, which could serve as markers of autophagy because they were involved in the formation of autophagosomes [
7,
60]. On the other hand, p62, an indicator of autophagic flux due to degradation in autolysosomes [
61,
62], was maintained at the control level. Therefore, phloroglucinol might protect ARPE-19 cells from H2O2-induced cellular damage, a pro-apoptotic mechanism, by counteracting the process of autophagy.



留言 (0)