
記住我



The concentrated granules (with a concentrate to dried herb ratio of 10:0.9) of four component herbs of BTWT, R. pulsatilla, R. coptidis, C. phellodendri, and C. fraxini (lot numbers 8055671, 9010401, 1612619, and 7122571, respectively), were purchased from Guangdong Yifang Pharmaceutical Co., Ltd (Foshan, China). The structures of representative components from the four herbs are shown in Fig. 1. AB4, AA3, 23-HA, aesculin, aesculetin, palmatine, jatrorrhizine, coptisine, epiberberine, and phellodendrine were purchased from Yuanye Bio-Technology Co., Ltd at the highest available purity (98%) (Shanghai, China). Berberine hydrochloride was purchased from Shenzhen ChemStrong Scientific Co., Ltd (Shenzhen, China) at the highest available purity (95%). DSS was purchased from MP Biomedicals (MW; 36,000 − 50,000, Solon, OH, USA). The stool bleeding test kit was purchased from BaSO Bio-Technology Co., Ltd (Zhuhai, China). Lipopolysaccharide (LPS) was purchased from Sigma-Aldrich. Deionized water was purified using a Millipore water purification system (Millipore, Milford, MA, USA). All other reagents used were of analytical grade.
Fig. 1
The label components of the four component herbs of BTWT
Analysis of the components of BTWT granules by liquid chromatography-mass spectrometry (LC–MS)Granules of the four herbs were accurately weighed (35.1 mg R. pulsatilla, 26 mg R. coptidis, 26 mg C. phellodendri and 15.6 mg C. fraxini) and dissolved in 2 mL of sterile water using an ultrasonicator. The final concentration of BTWT solution was 51.35 mg/ml. The solution was freshly prepared before analysis. For sample extraction, 0.59 ml methanol was added to 10 μl BTWT solution and vortexed for 10 min before centrifugation and evaporation. The residue was reconstituted in 1 ml mobile phase. The concentration of the sample for LC–MS detection was 513.5 μg/ml. A mixture of standard stock solution containing the 11 compounds was prepared in methanol at a final concentration of 3000 ng/ml AB4, 300 ng/ml AA3, 300 ng/ml 23-HA, 1200 ng/ml berberine, 1200 ng/ml palmatine, 1200 ng/ml coptisine, 800 ng/ml jateorhizine, 1500 ng/ml epiberberine, 300 ng/ml phellodendrine, 900 ng/ml aesculin, and 450 ng/ml aesculetin. Quantification was carried out on an ultra-high-performance liquid chromatograph (UPLC) with triple quadrupole tandem mass spectrometer (MS). The UPLC analysis was performed using a Shimadzu Nexera X2 LC-30AD (Shimadzu, Japan) system coupled to an AB Sciex 4500 QTRAP mass spectrometer (AB Sciex, USA) equipped with an electrospray ionization interface (ESI). Chromatographic separation was achieved on a Kinetex C18 column (Phenomenex, USA) (2.1 mm i.d. × 100 mm, 2.6 μm) equipped with a column heater. The mobile phase consisted of solvent A (0.1%, v/v, of formic acid in water) and solvent B (acetonitrile). The flow rate was 0.3 ml/min, the injection volume was 4 μL, and the column temperature was maintained at 35 ℃. Mass spectrometry data were acquired in multiple reaction monitoring mode and negative ESI mode.
Experimental animalsMale C57BL/6 mice, 8 –11 weeks old, weighing 23–25 g, obtained from Hunan Silaike Jingda Laboratory Animal Co Ltd (Hunan, China) were used in this study. The animals were housed at a controlled temperature (24 ± 2 °C) and with 12 h/12 h light/dark cycles. They were fed standard mice chow pellets and had access to filtered water. All animal care and experimental procedures were approved by the Animal Care and Use Committee at South China University of Technology (SCUT) (approval number: 2017004).
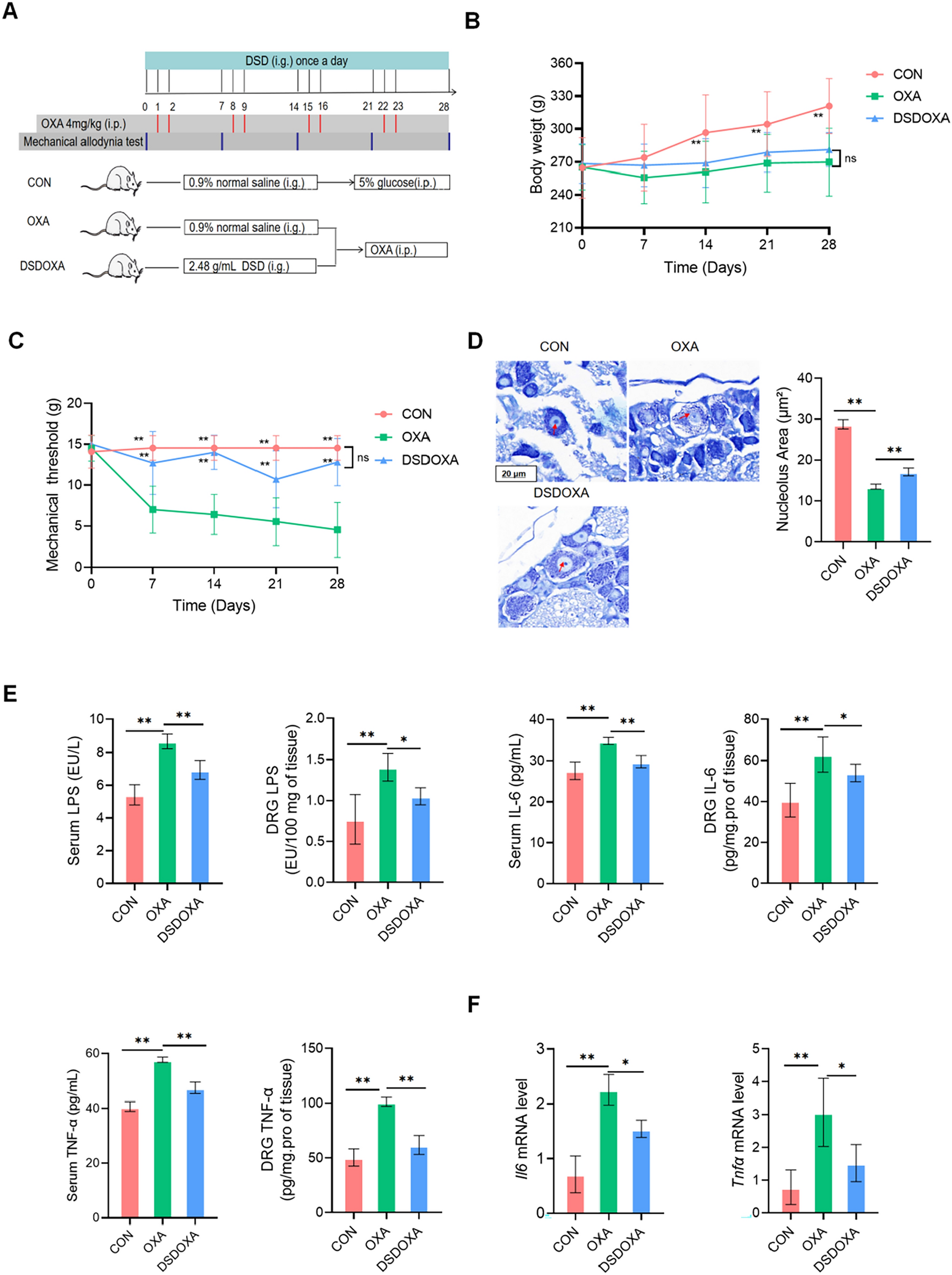
Induction of colitis and drug treatmentAcute colitis was induced by giving mice 2% DSS for 5 days and followed by 3 days of water. Chronic colitis was induced by giving mice 3 cycles of 2% DSS following a protocol previously described [27]. After 5 days or the first round of 4 days DSS administration, all mice developed symptoms of colitis and were randomly divided into two groups with five mice in each group; one group that received DSS-only, and one group that received BTWT (531.3 mg/kg). The dose equivalent to BTWTs granule was determined according to the human clinical equivalent dose of crude herb (45 g/70 kg for humans) and converted to 5.85 g/kg of crude herb for mice, and finally calculated as 513.5 mg/kg concentrated granules. All drug solutions were freshly prepared daily and orally administrated based on the actual weight of the mice (0.1 ml/10 g). The administration of drugs started on day 0 and lasted for 8 days for the acute model. For the chronic model, drug administration started on day 5 and lasted for 14 days. One group of mice designated as the water group received filtered water alone throughout the acute or chronic experimental procedure.
Determination of clinical score, colon length, and colon weightBody weight, stool consistency, and occurrence of occult blood or the presence of gross blood per rectum were determined according to previously described criteria [27,28,29]. Weight loss, stool consistency, and stool bleeding sub-scores were added, resulting in a total clinical score ranging from 0 (healthy) to 8 (maximal activity of colitis). At the end of treatment, the animals were sacrificed by cervical dissociation, and the entire colon from the cecum to the anus was removed. The colon length and colon weight were measured before the colon was divided into several segments for later biochemical examination.
Analysis of absorbed BTWT ingredients by LC–MS analysisPlasma (200 μL) and colon (70 mg) samples were taken from mice with acute colitis 1 h after the last intragastric administration of BTWT (531.5 mg/kg per day for 8 days), and the distribution of the 11 BTWT ingredients in these samples were determined by LC–MS analysis as described above.
Simulation setup and docking parametersAll in-silico simulations were executed using the software packages ChemDraw (version 16.0.082) and Molecular Operating Environment (MOE, version 2010.10). Seven compounds that are reported as respective ligands of the six selected protein targets were used as standards (positive controls). These compounds were as follows: the JAK3 inhibitor tofacitinib [30]; SD-36, which is a small-molecule degrader of STAT3 [31]; the S1PR1 endogenous ligand S1P [32] and the S1PR1 antagonist ML056 (W146); ML385, which blocks the activity of Nrf2 [33]; nivolumab, which is a monoclonal antibody against PD-1 [34]; and BSM-202, which is a small-molecule compound that potently blocks binding of PD-L1 to PD-1 [35]. The chemical structures of these positive-control ligands and selected components of BTWT were constructed using ChemDraw then imported into the MOE database file after 3-D protonation and energy minimization with the MMFF94x force field using default parameters.
The positive-control ligands for the docking simulations were created using MOE Builder and manually inserted into the active site of the six proteins extracted from the respective PDB files. The docking experiments were performed 100 times for each compound, and the 30 lowest energy conformations (poses) were recorded in order of increasing potential energy of the conformation in kcal/mol. For specific binding analysis, the best conformation for each compound was visualized and evaluated for analyses of binding modes between atoms or moieties of the ligands and specific residues of the target proteins [36].
Splenocyte culture and cell viability assaySplenocyteS isolated from the spleen of mice were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and 100 U/ml each of penicillin, and streptomycin (Gibico, Invitrogen, Carlsbad, CA, USA) overnight. To evaluate the cell viability, 2 × 105 splenocytes cultured in 96-well plates were treated with 1 µM of 11 components of BTWT for 24 h and then submitted to cell viability assay by mitochondria-dependent reduction of thiazolyl blue tetrazolium bromide (MTT) (Sigma-Aldrich). All experiments were repeated in quadruplicate in different intervals.
RAW 264.7 cell culture and drug treatmentRAW 264.7 cells were used as an in vitro model to test the effects of the 11 adsorbed BTWT compounds on cytokine production. RAW 264.7 cells were cultured at 1.0 × 106 cells/ml in 96-well plates in RPMI-1640 media (Gibco, Carlsbad, CA USA) containing 10% FBS (Gibco, Carlsbad, CA USA. Cells were pretreated with 1 μM tofacitinib, AB4, AA3, 23-HA, berberine, epiberberine, palmatine, coptisine, jateorhizine, phellodendrine, aesculin, or aesculetin for 22 h, and then incubated with 1 μg/ml LPS for 2 h at 37 °C in a humidified atmosphere with 5% CO2. At the end of the treatment, the supernatant from RAW 264.7 cell culture was collected and stored frozen (−80 °C) for analysis by enzyme-linked immunosorbent assay (ELISA) assay.
mRNA extraction and quantitative real-time reverse-transcription polymerase chain reaction (RT-qPCR)Total RNA from the colon or RAW 264.7 cells was isolated using TRIzol reagent (Invitrogen; ThermoFisher Scientific, Waltham, MA, USA) and subjected to mRNA purification using lithium chloride precipitation as previously described [28]. Reverse transcription was performed with a cDNA synthesis kit (TaKaRa Biotech, Japan). The mRNA expression of interleukin-17 (IL-17), interleukin-6 (IL-6), tumor necrosis factor alpha (TNF-α), and the internal control glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was measured by RT-qPCR using the 7500 real-time PCR system (Applied Biosystems; ThermoFisher Scientific, Waltham, MA, USA). Mouse mRNA primer sequences were the same as those used in our previous study [29]. The mRNA expression level in the water-treated control group was set as 1, and the mRNA expression levels in the drug-treated mice and cells were compared with those of the water-treated group.
ELISAThe levels of TNF-α and IL-6 in the cell culture supernatant of RAW 264.7 cells and splenocytes were measured using mouse Quantikine ELISA kits (TNF-α: Cat. No. 430904, IL-6: Cat. No. 431304, Biolegend, San Diego, CA, USA) according to the manufacturer’s protocol. All samples were analyzed in duplicates. The concentrations of TNF-α and IL-6 were calculated by standard curves and expressed as pg/ml.
Western blottingRAW 264.7 cells were homogenized in RIPA buffer and the protein concentration was determined using a BCA protein assay kit (Biosharp, Anhui, China). The immunoblot was then incubated with primary antibodies against phospho-STAT3 (pSTAT3; Cell Signaling Technology, Danvers, MA, USA; catalog number 9145 s), STAT3 (Proteintech, Rosemont, IL, USA; catalog number 60199–1-Ig), and β-actin (Affinity Biosciences, Jiangsu, China; catalog number T002-50). The obtained chemiluminescence signals were analyzed with ImageJ software.
Data analysisAll data are presented as the means ± standard deviation (SD) or means ± standard error of the mean (SEM) of at least three biological replicates. One-way or two-way ANOVA coupled with Dunnett’s post-test was used to test for statistically significant differences in the mean values between each treatment. Statistical analyses were performed using Prism 6 software for Windows (GraphPad Software, Inc., San Diego, CA, USA). P-values of 0.05 or less were considered statistically significant.
留言 (0)