
記住我

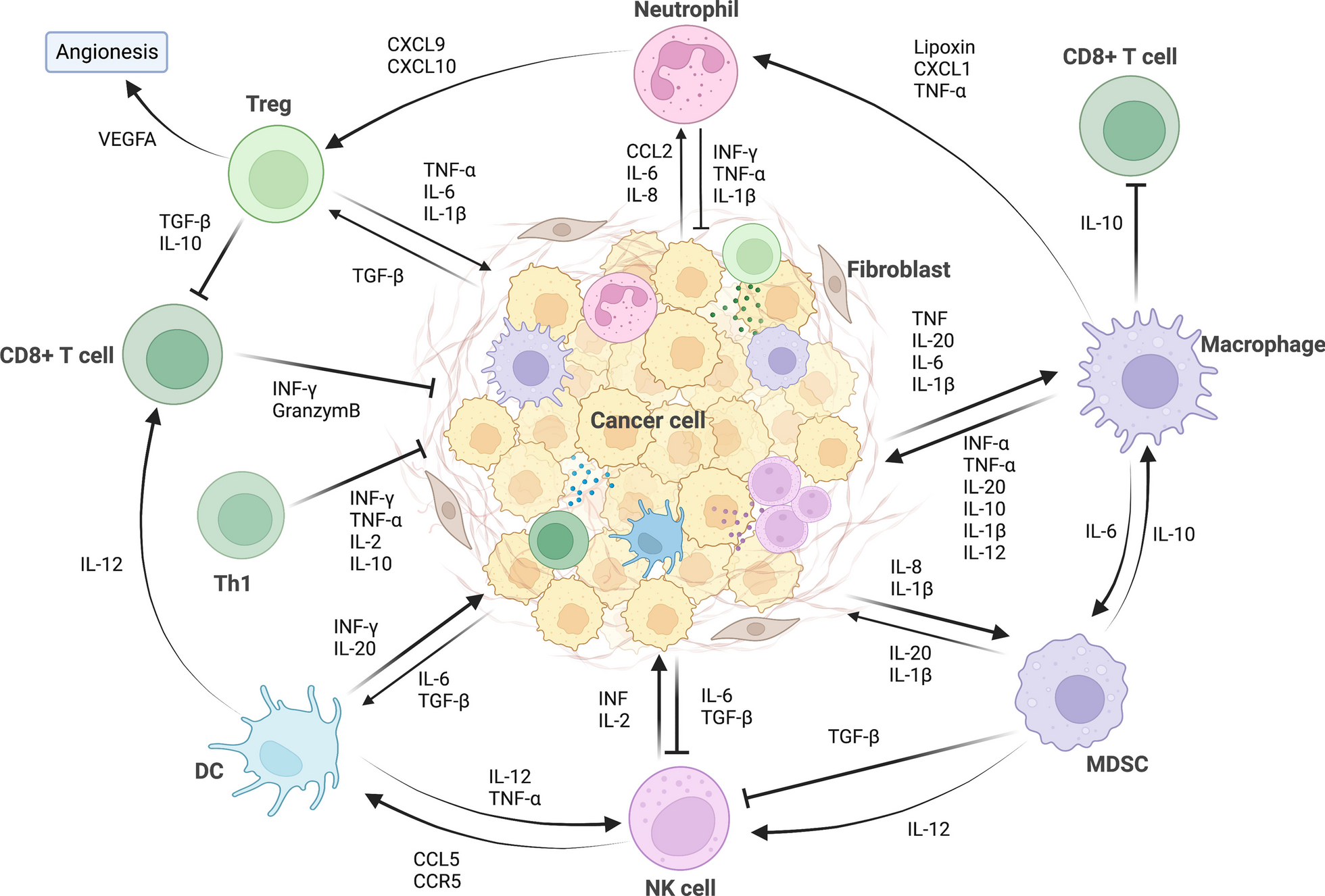


To characterize the proteogenomic landscape of pancreatic ductal adenocarcinoma (PDAC), whole-exome sequencing (WES), RNA-seq, proteomic, and phosphorylation proteomic data were collected from 229 treatment-naive patients (Additional file 23: Table S1A). HE-stained slides were examined and evaluated independently by two experienced pathologists and information regarding tumor histological subtype, degree of differentiation, TNM stage, and tumor purity were provided (Methods). All formalin-fixed paraffin-embedded (FFPE) samples used in this study had tumor purity ranged from 10 to 80% (median 50%) (Additional file 23: Table S1B). Neoplastic cellularity was evaluated independently by whole-exome sequencing using the ABSOLUTE [79] and ESTIMATE [78] algorithm (Methods) and ranged from 10 to 60% (median 36%), 1% to 70% (median 50%), respectively (Additional file 23: Table S1B). Computational purity showed significantly positive correlation with our histological evaluated tumor purity (Spearman’s = 0.98 for ABSOLUTE and 0.91 for ESTIMATE, P < 0.0001) (Additional file 19: Fig. S19A). A schematic of the experimental design is shown in Fig. 1A, and the clinical metadata are shown in Additional file 23: Table S1B. Clinical data, including the gender, age at diagnosis, tumor grade, and survival, etc., are summarized in Table 1. Comparing to recently published PDAC dataset conducted by CPTAC [17], our cohort showed distinctive demographic and clinical characteristics. Demographically, all the patients in our cohort were from Asian (Asian, n = 229, 100%), whereas 21 patients in CPTAC cohort were from Asian (Asian, n = 21, 15%); histologically, more early-stage patients (our cohort: stage IA/IB, n = 105, 46%, CPTAC cohort: stage IA/IB, n = 23, 16%; fisher exact test, P < 1.0E−4) were included in our cohort, which facilitated us to investigate the specific molecular features of early-stage PDAC for tumor diagnosis. Additionally, other risk factors and information associated with patients’ prognosis, such as metastasis status and diabetes, were also collected via follow-up in our study (Fig. 1A, Table 1 and Additional file 1: S1B). Proteomic profiling was performed on the 226 tumor and 220 non-tumor adjacent tissues (NATs). The WES analysis was conducted on the 149 paired samples, and the phosphoproteomic analysis was performed on the 113 paired samples, respectively. In addition, the mRNA sequencing was carried out on the 54 tumors and 51 paired NATs. Therefore, our study provided a comprehensive landscape of PDAC at the multi-omics level.
Fig. 1
Multi-omics landscape of PDAC Samples. A Overview of the experimental design and the number of samples for the genomic, transcriptomic, proteomic, and phosphoproteomic analyses. B The genomic profiles of PDAC. The top 20 mutated genes and their occurrence in 149 PDAC patients and the mutation frequencies are shown. Mutation types and their frequencies are demonstrated by a bar plot in the right panel. C Spearman’s correlation plot indicating the mutation frequencies observed in the Fudan cohort compared to previously published cohorts. D Comparisons of mutation frequencies of top 10 mutated genes in the Fudan cohort and previously published cohorts. E Bar plots of the common KRAS driver mutations in Fudan cohort and previously published cohorts. ****p < 1.0E−4, ***p < 1.0E−3, **p < 1.0E−2, *p < 0.05, ns > 0.05
Table 1 Demographic characteristics of the patients in Fudan cohort and CPTAC cohortWES profiling identified 19,530 genetic variation events (14,656 missense, 869 nonsense, 1976 in-frame, and 2029 frame-shift mutations). Significantly mutated genes (SMGs) were determined using OncodriveCLUST (Methods) [18], and mutations, such as KRAS (95%), TP53 (49%), TTN (32%), MUC16 (23%), CDKN2A (18%), and SMAD4 (17%), were identified as the top-ranked mutations, in our cohort (Fig. 1B). Correlation analysis using mutational frequencies from other pancreatic carcinoma (PC) cohorts, including TCGA cohort [11], UTSW cohort [12], ICGC cohort [13, 14], and CPTAC cohort [17], resulted in an average of Spearman’s correlation coefficient was 0.8 among different cohorts, in which no significant difference was observed (Fig. 1C, Additional file 24: Table S2C-H). In addition, the mutational frequencies of KRAS, TP53, CDKN2A, and SMAD4 were 95%, 49%, 18%, and 17% in our cohort, respectively, which showed similarity across the different cohorts (Fig. 1D). For KRAS mutations, except for the most common mutations of KRAS G12 (90.3%), we also identified the other KRAS codon mutations KRAS (G13D), KRAS (Q61H) (Fig. 1E).
The frequencies of mutated trinucleotide sequence motifs were analyzed using nonnegative matrix factorization (NMF) [19, 20] (Methods). Moreover, cosine similarity analysis against mutational signatures in human cancer was performed to reveal the potential contribution of endogenous and exogenous mutagens in PDAC, and five mutational profiles were identified. As a result, the mutational signatures best matching to those in our patients was SBS1, SBS 6 and SBS30 (Additional file 1: Fig. S1E). The same enrichment analysis was performed in the CPTAC cohort [17]. As a result, the mutational signatures best matching to those in CPTAC patients was SBS 1 and 6, associated with patients age at diagnosis (SBS1), and tumor mutation burden (SBS6) (Additional file 1: Fig. S1E and F).
As for proteomic analysis, 16,584 proteins were identified, with 9006 proteins per sample on average (Additional file 1: Fig. S1H-J) (Methods). Whole cell extract of HEK293T cells was used as quality control (QC) for mass spectrometers. This extract showed the robustness and consistency of the mass spectrometer, which was evidenced by a high Spearman’s correlation coefficient (r > 0.88) between the proteomes of QC samples (Additional file 1: Fig. S1G). Principal component analysis of proteomic data was conducted based on years of sample collection and experimental batches and found no obvious distinction, indicating that there was no batch effect (Additional file 1: Fig. S1L). Further, 15,443 and 15,092 proteins were identified in the tumors and NATs, respectively (Additional file 1: Fig. S1B). A total of 18,883 and 17,098 transcripts were identified in the tumors and NATs, giving an opportunity to probe the relationship between the transcriptome and proteome (Additional file 1: Fig. S1A).
Phosphoproteomic analysis identified 33,426 phosphosites including 24,470 (73.2%) on serine, 7914 (23.7%) on threonine, and 1042 (3.1%) on tyrosine, from 6153 phosphoproteins in 113 tumors; 33,696 phosphosites, including 24,721 (73.4%) on serine, 7965 (23.6%) on threonine, and 1010 (3.0%) on tyrosine, from 6243 phosphoproteins in 113 NATs (Additional file 1: Fig. S1C-D, Additional file 7: Fig. S7A). Comparing the phosphorylation sites S/T/Y distribution between HCC cohort [21], GC cohort [22], and CRC cohort [23], the ratio of S/T/Y in our cohort is similar to those cohorts (HCC cohort: 77.8%, 16.9%, and 5.3%, GC cohort:74.0%, 20.9%, and 5.1%, CRC cohort: 76.2%, 19.9%, and 3.9%), indicating that the ratio of S/T/Y in gastrointestinal tumors (including pancreatic cancer, liver cancer, gastric cancer, colon cancer, etc.) is comparable (Additional file 7: Fig. S7A-B). Importantly, the phosphorylation of some key molecules in tumor tissue was significantly enriched, providing an opportunity to explore key oncogenic phosphorylation modifications. For example, RB1 (retinoblastoma-associated protein) at S807, which could promote cell cycle progression in cancer cells [24], YAP1 at S109, which has been reported associated with tumor metastasis [25], was significantly overexpressed in tumor tissues. We further presented the MS2 spectrums of these phosphosites to verify the accuracy of our detection. Additionally, we utilized IHC (immunohistochemistry) with phosphorylation antibodies targeted to YAP1_pS109 and RB1_pS807 and confirmed their enrichment in tumor tissues (Additional file 2: Fig. S2A-C).
Thus, our study has so far established a comprehensive landscape of PDAC at the genomic, transcriptomic, proteomic, and phosphoproteomic levels (Additional file 1: Fig. S1K).
The impacts of somatic copy number alterations in PDAC cohortWe applied GISTIC2 [26] to analyze the somatic DNA copy-number profiles of 149 PDAC samples. The most frequent gains were identified in chromosomes 14p (q = 8.4E−5), 21p (q = 1.4E−6) and 22p (q = 3.5E−8), and the most frequent losses were observed in chromosome 21p (q = 1.1E−2) and 17q (q = 2.8E−2) (Fig. 2A, Methods). In addition, we identified amplifications in driver oncogenes such as AKT1 (14q32.33) and PDGFB (22q13.1) and deletions of the pancreas-specific genes such as PNLIP and PNLIPRP1 (10q25.3) (Fig. 2B, Additional file 24: Table S2A-B).
Fig. 2
The Impacts of Somatic Copy Number Alterations in PDAC Cohort. A Significant arm-level focal peaks detected using GISTIC. B Focal-level SCNA events. Focal peaks with significant copy-number gains (red) and losses (blue) (GISTIC2 FDR < 0.05) are shown. The focal peaks are highlighted in approximate positions across the genome. C Venn diagram depicting the process of screening for TFs with cis-effect between CNA and protein. D The heatmap showing the cis-effect of IRF6 among CNA, RNA, protein, and phosphosite (Spearman’s correlation). The P value of correlation is noted on the left. E Kaplan–Meier curves for overall survival based on mRNA abundance of IRF6 (left), and TF activity of IRF6 (right) in Fudan cohort (log-rank test). F Kaplan–Meier curves for overall survival based on mRNA abundance of IRF6 in TCGA-PAAD dataset (log-rank test). G Spearman-rank correlation of the abundance of IRF6 and its target genes (Spearman’s correlation). The significant correlations are colored in dark gray. Cell cycle and cell proliferation-associated proteins are labeled in pink, and prognostic related proteins are labeled with purple circles. H The scatter plot describing the Spearman’s correlation between IRF6 protein expression and GSVA score of cell proliferation. I Kaplan–Meier curves for overall survival based on GSVA scores of cell proliferation (log-rank test). J, K The scatter plot describing the Spearman’s correlation between MCM4 protein expression and MGPS score (J) or tumor size (K). L The systematic diagram summarizing the impact of the cis-effect of GRB7 amplification on increasing tumor size. M The boxplot revealing the comparison of GSVA score of cell proliferation between the younger patients (≤ 60) and the older patients (> 60) (Student’s t test). N The forest plot indicating the hazard ratio of KRAS, TP53, SMAD4, and CDKN2A, in younger patients (left) and older patients (right). O The pathway heatmap indicating the enriched pathways in the four groups (TP53 mut, younger patients; WT, younger patients; TP53 mut, older patients; WT, older patients). Each column represents a type of sample. The color of each cell shows -log10 transformed P value. P Venn diagram depicting the process of screening for highly expressed kinase in younger patients with TP53 mutations. Q The boxplot revealing the comparison of CDK4 protein expression between TP53 mutations and WT both in the younger patients (≤ 60) and the older patients (> 60) (Wilcoxon test). R Kaplan–Meier curves for overall survival based on CDK4 abundance in the younger patients (left) or the older patients (right) (log-rank test). S The heatmap showing cell cycle-associated phosphosites which are positively correlated with CDK4. T The systematic diagram summarizing the impact of TP53 mutations on promoting cell cycle. ****p < 1.0E−4, ***p < 1.0E−3, **p < 1.0E−2, *p < 0.05, ns > 0.05
The impacts of copy-number alterations (CNAs) on mRNA, protein, and phosphoprotein abundances in both cis or trans mode were characterized. In total, 1286 and 1277 significant positive correlations (cis/trans-effects) were observed for proteins and phosphoproteins, respectively (Spearman’s correlation, P < 0.05). The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis indicated consistency among pathways subjected to enrichment by CNA-affected proteins, and phosphoproteins in the same sample type. For instance, CNA-affected proteins and phosphoproteins were enriched in ECM–receptor interaction, RNA transport, etc. (Fisher’s exact test, P < 0.05) (Additional file 3: Fig. S3A).
To further nominate functionally important genes within CNA regions, we focused on the 551 cancer-associated genes (CAGs) [27]. A total of 17 significant positive correlations were observed for proteins (Spearman’s correlation, P < 0.05), including 3 transcription factors (TFs: IRF6, CIC, and FOXO3) (Fig. 2C). IRF6, the top-ranked gene, was significantly associated with its cognate RNA, protein, and phosphorylation site (Fig. 2D). Further survival analysis indicated both the mRNA expression and the inferred TF activity of IRF6 were significantly associated with unfavorable overall survival. This observation was further supported by TCGA data (Fig. 2E, F). IRF6 is a transcription factor that has been reported to participate in various cellular process through transcription regulation [28]. Ke Zhang et al. have reported that IRF6 might be a prognostic marker in pancreatic cancer [29]. To gain great insight into the mechanism of how IRF6 led to poor prognosis, we applied correlation analysis on the target genes (TGs) of IRF6. As a result, the TGs significantly associated with the protein expression IRF6 were mainly enriched in cell cycle and cell proliferation. The expression of TGs including MCM4, TTL12, EZR, and the GSVA scores of cell cycle was positively correlated with the protein expression of IRF6 (Fig. 2G–H). Moreover, survival analysis indicated in concordant with the expression of IRF6, the enhanced cell proliferation also led to poor prognosis (Fig. 2I). These results suggested IRF6 could promote the cell proliferation through transcriptional regulation. Since enhanced cell proliferation could be represented by the elevated MGPS (multi-gene proliferation score) and enhanced tumor size, we evaluated the association between IRF6’s TGs and both MGPS and tumor size (Methods). As a result, the most significant positive correlations were observed between MCM4 (TG of IRF6) with both MGPS (correlation = 0.47, P = 6.7E−14) and tumor size (correlation = 0.25, P = 0.04), emphasized the important role of MCM4 in IRF6-mediated cell proliferation (Fig. 2J, K). In sum, our data revealed a potential mechanism that the cis-effect of IRF6 promoted the progression of PDAC by influencing cell cycle process (Fig. 2L).
To explore other risk factors which might associate with tumor cell proliferation in PDAC, we further investigated the effect of diverse clinical characteristics (age, gender, etc.) on cell proliferation. PDAC is considered a disease of the elderly, with patients mostly over the age of 65 [30]. Individuals diagnosed with PDAC under the age of 60 are considered to be young-onset and potentially at high risk for a genetic predisposition. Intriguingly, we observed that the younger patients (age at diagnosis ≤ 60) in our cohort showed elevated GSVA scores of cell proliferation (Fig. 2M). To elucidate the possible causes and impacts of this phenomenon, we first compared the SCNAs, RNA, and protein expression of IRF6 between older and younger patients (older: > 60, younger: ≤ 60) and observed no significant difference (Additional file 3: Fig. S3B). Survival analysis indicated the elevated expression of IRF6 associated with poor prognosis in both older and younger patients (Additional file 3: Fig. S3C). We then compared the prognostic relevance of SMGs between the two groups and observed the mutations of TP53 only impacted the overall survival in patients younger than 60 (Fig. 2N). Comparative analysis between TP53-mutant and TP53-wild-type patients, in the two age groups, further illustrated that in younger patient group, the cell cycle process was elevated in TP53-mutant patients, whereas, in older patient group, the cell cycle process was dominant in TP53-wild-type patients (Fig. 2O). To further illustrate the mechanism, underline how TP53 mutations impact the cell cycle process, we performed comparative analysis and identified 7 cell cycle-related proteins (RB1, PRKDC, CDK4, MCM4, SMC1A, SKP1, and ANAPC2) which showed elevated expression in younger TP53-mutant patients (Fig. 2P, Q, Additional file 3: Fig. S3D). Further survival analysis revealed that the CDK4 was significantly associated with poor prognosis only in younger TP53-mutant patients (log-rank t test, P < 0.05) (Fig. 2R). To illustrate the impact of CDK4 on kinase–substrate phosphorylation, we screened out phosphosubstrates regulated by referred kinases from public database [31] and calculated the correlation between the abundance of these phosphosubstrates and the protein expression of CDK4. As a result, the abundance of phosphosites enriched in cell cycle such as CDK11A_pS268, CDK17_pS900, and EP300_pS900 were significantly associated with the expression of CDK4 (Fig. 2S, Additional file 3: Fig. S3E), suggesting that the CDK4 regulated the cell cycle process through phosphorylation signaling pathway. Together, this result revealed the role of TP53 mutations in promoting tumor cell proliferation, through CDK4-mediated signaling pathway in younger patients, and suggested promising therapeutical options of younger TP53-mutant patients by CDK4 inhibitors (Fig. 2T).
Phosphorylation of transcription factor E2F1 promoting the tumor cell proliferation feature of patients with KRASG12D mutationsKRAS is the well-known oncogene with the highest mutation rates in PDAC. Unfortunately, there are still no effective strategies targeting KRAS mutations. Due to the intrinsic characteristics of KRAS protein, targeting KRAS has been quite challenging [32]. Therefore, many efforts have focused on targeting it downstream signaling effectors.
To investigate signal transduction pathways downstream of activated KRAS in search of alternative therapeutic targets, we evaluated phosphosite expression changes in tumors with different KRAS hotspot mutations. As a result, comparing to tumors with KRASG12R, KRASG12V, KRASQ61H, and KRASQ61R mutations, tumors with KRASG12D mutations harbored elevated phosphorylation of RAF1 at Ser 259, MAP2K2 at Tyr 222, MAPK1 at Ser188, and E2F1 at Ser 364, which indicated the elevation of RAS-RAF-MEK-ERK-E2F1 phosphorylation cascade in tumors with KRASG12D mutations, and E2F1 at Ser 364 had the most significant elevation among these phosphorylation sites (Additional file 4: Fig. S4A, B).
Functionally, E2F1, as a cell cycle-specific transcription factor (TF), has been reported to be highly expressed in a variety of tumors, and mainly participated in regulating tumor cell proliferation [33, 34]. To evaluate the prognostic value of elevated phosphorylation of E2F1, we conducted survival analysis and found that the elevated phosphorylation of E2F1 at Ser 364 was negatively associated with patients’ overall survival (Additional file 4: Fig. S4C), which emphasized its therapeutical potential in the future.
To further investigate the mechanism that how the elevated phosphorylation of E2F1 led to poor prognosis, we inferred the TF activity of E2F1 based on GSVA algorism (Methods) and found that along with the phosphorylation of E2F1 at Ser 364, its TF activity was also negatively associated with patients’ overall survival (Additional file 4: Fig. S4C). Intriguingly, the protein expression of E2F1 showed no difference among tumors with different KRAS hotspot mutations (Additional file 4: Fig. S4D). In supporting of the observation, we also conducted IHC staining utilizing both E2F1_pS364 antibody and E2F1 antibody and found only the phosphorylation of E2F1 showed significant elevation in tumors with KRASG12D mutations (Additional file 4: Fig. S4E). Consistently, the inferred TF activity of E2F1 showed high correlation with the abundance of phosphorylation of E2F1 at S364 but no correlation with the E2F1 protein expression (Additional file 4: Fig. S4F). These findings indicated the TF activity of E2F1 was contributed by phospho-E2F1 rather than the abundance of E2F1 protein.
We then hypothesized that the phosphorylation of E2F1 might impact prognosis in patients with KRASG12D, through transcription regulation. Along with this hypothesis, we surveyed the expression of E2F1’s TGs and identified that the mRNA expression of TGs including AHCTF1, CDC27, ATM, TAOK3, RB1, and CDK14, which enriched in cell cycle process, was most significantly associated with the TF activity of E2F1 (Additional file 4: Fig. S4F). Interestingly, further correlation analysis revealed the significantly positive correlation between those TGs’ mRNA expression and their cognate protein expression, which revealed that the transcriptional regulatory pattern led by E2F1 was inherited at protein level (Additional file 4: Fig. S4G). We then performed survival analysis and observed the protein expression of TGs including AHCTF1, CDC27, MAP4K5, etc., which were also negatively associated with patients’ overall survival (Additional file 4: Fig. S4H).
In sum, our finding revealed the phosphorylation of E2F1 at S364, but not protein expression, elevated its TF activity which enhanced the cell proliferation process, and led to poor prognosis in tumors with KRASG12D mutations (Additional file 4: Fig. S4I).
Integrated multi-omics features in tumor tissues compared with NATs of the PDACMulti-dimensional omics data provided an excellent chance to explore the relationships between the transcriptome, proteome, and phosphoproteome of PDAC. After appropriate sample quality control (QC) and normalization procedures (Methods), we performed integrated multi-omics analysis to systematically represent the characteristics of PDAC. Principal component analysis (PCA) of transcriptome, proteome, and phosphoproteome data was conducted to demonstrate a clear distinction between tumors and NATs, which further highlighted the diverse expression patterns existing between pancreatic tumor tissues and normal adjacent tissues that emphasized our stratification analysis (Fig. 3A, Additional file 5: Fig. S5A-B).
Fig. 3
Integrated multi-omics features in tumor tissues Compared with NATs of the PDAC. A PCA of 7055 proteins in 226 tumors and 220 normal adjacent tissues (NATs). Red, tumors; blue, NATs. B A volcano plot showing the results of a two-tailed Student’s t test comparing tumors and NATs at proteomic level. The number of significantly increased and significantly decreased proteins in tumors is shown above the volcano plot. C Differentially expressed proteins and phosphoproteins in tumors and NATs. The enriched biological pathways are annotated on the right. D The expression of pancreatic signature proteins in tumors and NATs at multi-omics level (mRNA, protein, and phosphoprotein). E The scatter plot indicating the changes of protein and mRNA between tumors and NATs. Proteins are labeled based on the pathways they are enriched in (proteins participated in fatty acid degradation, pancreatic secretion, and protein processing and export are labeled in purple, light-blue, and green, respectively; protein participated in endocytosis and lysosome are labeled in red, and proteins participated in focal adhesion are labeled in orange). The systematic diagram summarizing proteins and signaling cascades that are significantly altered in NAT (protein processing and export, pancreatic secretion), and in tumor (lysosome, focal adhesion). Values are color coded based on the fold change between tumors and NATs at both transcriptomic and proteomic level. F The screening process of potential biomarkers in PDAC showing on the left. The expression heatmap describes the protein expression of potential biomarkers (middle). Color of each cell shows Z-scored average abundance of the protein across the tumor and NAT samples. The ratio of RNA expression between tumors and NATs is shown on the right, and the heatmap on the left indicates the fold change of proteins average expression between pancreatic cancer exosomes versus normal exosomes. Bar plot shows the ratio of protein and RNA expression of these biomarkers between early-stage patients (TNM IA and IB) and others (right). Proteins are labeled in blue, and RNAs are labeled in yellow. G KSEA analyses of kinase activities in tumors and NATs. H The volcano plot depicting the correlation between phosphosubstrates and the kinase activity of PRKCD; the significant correlations are colored in pink. I Spearman rank correlation of the abundance of phosphosite STAT1_pS727 and the kinase activity of PRKCD (Spearman’s correlation). J Heatmap of the relative abundance of focal-adhesion-related proteins (bottom panel) and STAT1 mRNA (top panel) that are significantly associated with TF activity of STAT1. The P value of survival is shown on the right. K The systematic diagram summarizing proteins and signaling cascades that are regulated by the kinase PRKCD. ****p < 1.0E−4, ***p < 1.0E−3, **p < 1.0E−2, *p < 0.05, ns > 0.05
To identify protein network associated with PDAC, we applied Student’s t test and identified 2136 (30%) and 762 (11%) proteins that were significantly overrepresented (FC > 2, Student’s t test, Benjamini–Hochberg adjusted p < 0.05) in the tumors, and NATs, respectively (Fig. 3B, Additional file 25: Table S3A). Further enrichment analysis of KEGG ontologies indicated that complement and coagulation cascades, focal adhesion, and platelet activation were significantly enriched in tumor tissues, whereas pathways related to previously reported pancreas physiology functions [35], including that amino acid metabolism, pancreatic secretion, protein processing, and export were enriched in NATs (Fig. 3C). The consistency of the dominant pathways enriched in tumors or in NATs was also observed at the transcriptome and phosphoproteome levels (Additional file 5: Fig. S5C-G, Additional file 25: Table S3B). This distinctive proteomic features of tumor and NATs were further supported by CPATC data [17]. To be more specific, 1918 proteins (56%) were identified to be upregulated and 963 proteins (72%) to be downregulated in both our cohort and CPTAC cohort, respectively (Additional file 6: Fig. S6A). Proteins enriched in ECM–receptor interaction and focal adhesion were significantly elevated in tumor tissues, while proteins enriched in pancreatic secretion, protein digestion, and absorption were significantly elevated in NATs (P < 0.05) in both CPTAC and Fudan cohort (Additional file 5: Fig. S5F and Additional file 6: Fig. S6B).
To understand the regulatory relationship among multi-omics, we calculated gene-wise (inter-sample) and sample-wise (intra-sample) correlation. Between transcriptome and proteome, NATs displayed a median gene-wise correlation value of 0.2, while tumors displayed a higher median value of 0.32 (Additional file 5: Fig. S5H-I). For PDAC tumors and NATs, 24% and 14% of mRNA-protein pairs had significant positive Spearman’s correlations, respectively (Additional file 5: Fig. S5H-I, Benjamini–Hochberg adjusted p < 0.05), with DNA repair, G2M checkpoint and lysosome, etc., positively correlated in tumors. In NATs, the metabolic process, such as oxidative phosphorylation, glycolysis, and pancreatic secretion, displayed a positive correlation pattern, representing the coherence of mRNAs and proteins in regulating the key pathways in tumor and NATs. In addition, although proteins participated in focal adhesion, lysosome, pancreatic secretion, and protein processing and export showed significant alteration between tumors and tumor-adjacent tissues both at proteome level and transcriptome level. The data showed more significant alteration between tumors and NATs at proteome level, highlighting the existence of a differentially regulated axis between transcriptome and proteome for the maintenance of carcinogenesis (Fig. 3E, Additional file 25: Table S3C). The significantly altered pathways and mapped key proteins are summarized in Fig. 3F.
Further correlation analysis between proteome and phosphoproteome showed that NATs displayed a median gene-wise correlation value of 0.12, while tumors displayed a higher median value of 0.26 (Additional file 5: Fig. S5J-K). For tumors and NATs, 14.7% and 32.3% proteins-phosphoprotein pairs had significant positive Spearman’s correlations, respectively (Additional file 5: Fig. S5J-K, Benjamini–Hochberg adjusted p < 0.05), with lipid metabolism, glycolysis, and pancreatic secretion, showed positive correlation in NATs. In tumors, the process that have been reported in PDAC tumorigenesis and metastasis such as cell cycle process, P53 pathway and WNT pathway displayed a positive correlation pattern, further revealed the concordance among different omics layer in regulating core process in tumors and NATs.
We further focused on the expression of pancreatic markers between tumors and NATs based on the human protein atlas database (https://www.proteinatlas.org/). Importantly, 68.5% (183/267) of detected pancreas-specific proteins, which mainly enriched in pancreatic-specific metabolic pathways such as pancreatic secretion, protein digestion and absorption, and fat digestion and absorption, were significantly attenuated in the tumors (Fig. S6C). Survival analysis indicated that 43.7% (80/183) showed positive association with overall survival. In addition, the proteins which had the highest level of enriched expression in pancreas (including CELA2A, CPA1, PRSS1 etc.) and specific proteins of ductal cells of pancreas (including CFTR and DCDC2) showed decreased expression at multi-omics level (Fig. 3D). Concordantly, the pancreas-specific proteins including CELA2A, PRSS1, and PNLIPRP1 were also detected to be enriched in NATs, in the CPTAC cohort [17], confirming the loss of pancreas functions could reflect the malignant of the PDAC (Additional file 6: Fig. S6D).
Our dataset provided a good opportunity to identify potential prognostic biomarkers and drug target of PDAC. We hypothesized that the most highly and commonly expressed proteins with prognostic power in tumor could be circulated and detected in body fluid or exosome samples, and could be used as potential biomarkers. Under this assumption, we performed differential gene expression analysis among 1416 proteins expressed in all the 226 tumor samples and identified 349 significantly altered proteins (FC > 2, P < 0.05), in which 15 proteins were negatively correlated with the overall survival. These 15 molecules were also significantly upregulated in tumors at transcriptome level. To further validate the prognostic value of the 15 proteomic biomarkers, we included proteomic and clinical data from CPTAC PDAC study, compared the protein expression level of these 15 proteins between tumors and NATs, and evaluated their relevance with patients’ overall survival. As a result, in concordant with our result, all 15 proteins showed elevated expression in tumors in CPATC cohort, and 14 of them were negatively associated with patients’ overall survival (Fig.
留言 (0)