
記住我








Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has caused a global health emergency, with many people developing clinically significant COVID-19, and 20% requiring hospitalization (1-3). One third of hospitalized patients develop acute respiratory distress syndrome (ARDS), a life-threatening inflammatory lung condition characterized by loss of aerated tissue, severe hypoxemia, and increased dead space (4,5). Despite standard use of lung-protective ventilation strategies and prone positioning, patients with COVID-19–associated ARDS requiring invasive mechanical ventilation have poor outcomes (4,6–8). Among these patients, only dexamethasone plus interleukin (IL)-6 inhibition or Janus kinase (JAK) inhibition has been shown to improve survival, but mortality remains high (9-11). Therefore, an unmet need remains for additional effective therapies.
COVID-19 is associated with aberrant cytokine signaling, including overactivation of the JAK/signal transducers and activators of transcription (STAT) pathway (12). Several cytokines that activate JAK/STAT signaling, including interferon-γ, granulocyte-macrophage colony-stimulating factor, IL-2, and IL-6, are overexpressed in patients with COVID-19 (13,14), and inhibiting some of these inflammatory mediators is associated with clinical improvement compared with standard of care (10,15,16). The IL-6 inhibitor tocilizumab has received emergency use authorization; however, evidence from randomized trials of patients with severe disease is mixed (10,17). Because the COVID-19–associated cytokine storm involves several cytokines in addition to IL-6, blocking multiple inflammatory mediators via JAK/STAT inhibition is a rational therapeutic approach (12). Furthermore, the inflammatory response of COVID-19 exhibits a similar macrophage-derived cytokine profile as hemophagocytic lymphohistiocytosis (18), which is responsive to the selective JAK1/JAK2 inhibitor ruxolitinib (19,20). Several reports have provided proof of concept for JAK inhibition for the treatment of critically ill patients with COVID-19 (11,21-24); however, only one exploratory study has investigated a JAK inhibitor (baricitinib) specifically in patients with COVID-19–associated ARDS requiring mechanical ventilation (11). The objective of this study was to evaluate the effects on mortality and in-hospital outcomes (ventilator-, ICU-, oxygen-, vasopressor-, and hospital-free days) as well as the safety of the investigational agent ruxolitinib in mechanically ventilated patients with COVID-19–associated ARDS.
METHODS Study Design and PatientsRuxolitinib in Participants With COVID-19–Associated ARDS Who Require Mechanical Ventilation (RUXCOVID-DEVENT; ClinicalTrials.gov‚ NCT04377620) was a double-blind, randomized, placebo-controlled, multicenter phase 3 trial assessing ruxolitinib in mechanically ventilated patients with COVID-19–associated ARDS. Eligible patients were at least 12 years old, hospitalized with SARS-CoV-2 infection (confirmed ≤ 3 wk before randomization), mechanically ventilated with arterial oxygen partial pressure/fractional inspired oxygen (Pao2/Fio2) of less than or equal to 300 mm Hg within 6 hours of randomization, and had bilateral pulmonary infiltrates on x-ray or CT chest scan. Exclusion criteria included suspected active uncontrolled bacterial, fungal, viral, or other infection (besides COVID-19); being unlikely to survive longer than 24 hours from randomization per investigator judgment; receiving extracorporeal membrane oxygenation; or receiving JAK inhibitor treatment within 30 days of randomization.
The central institutional review board (WCG Western Institutional Review Board) approved the study (approval number 20201074). Informed consent was obtained from patients’ healthcare proxy per an institutionally approved method before study treatment initiation. The trial was performed in accordance with the principles embodied by the Declaration of Helsinki with adherence to the trial protocol and the International Council for Harmonisation Guideline for Good Clinical Practice.
ProceduresInteractive response technology was used to block randomize patients 2:2:1 to ruxolitinib 15 mg twice a day (BID)‚ ruxolitinib 5 mg BID‚ or matching dual-dose placebo tablets. Given the expected high mortality rate and anticipation of a treatment effect, the 2:2:1 ratio was employed to allow for ~80% of patients to receive ruxolitinib. Randomization was stratified by ARDS severity (mild/moderate [Pao2/Fio2 > 100–300 mm Hg] vs severe [Pao2/Fio2 ≤ 100 mm Hg]) and investigational site. Treatment identity was concealed by the use of identical packaging, administration schedule, appearance, taste, and color. The study statistician and programming team were unblinded before database lock (i.e., no further changes to trial data allowed) after statistical analysis plan finalization to confirm that randomization codes had been applied correctly by the independent statistician; otherwise, patients, investigators, and the study sponsor remained blinded to treatment from the time of randomization until database lock. A 1-day screening period was followed by a 28-day follow-up period (eFig. 1, https://links.lww.com/CCM/H210). The initial treatment period lasted 14 days, after which patients could continue to receive ruxolitinib or placebo treatment for an additional 14 days if the benefit and risk were deemed appropriate in the opinion of the site investigator in consultation with the blinded medical monitor on Day 15, without knowledge of study drug identity. Crossover between groups was not permitted. Safety follow-up assessments were conducted 28 ± 3 days after the last dose of trial treatment. Trial treatment was administered via an enteric feeding tube or orally, as clinically appropriate.
OutcomesThe primary endpoint was 28-day mortality, defined as the proportion of patients who died due to any cause through Day 29. Secondary efficacy endpoints included in-hospital outcomes (number of ventilator-free days, ICU-free days, oxygen-free days, vasopressor-free days, and hospital-free days) at day 29; change in Sequential Organ Failure Assessment (SOFA) score from baseline to days 3, 5, 8, 11, 15, and 29; and clinical status using the World Health Organization (WHO) nine-point ordinal scale at days 15 and 29 (eTable 1, https://links.lww.com/CCM/H210) (25). The safety endpoint was the number and proportion of patients with treatment-emergent adverse events (TEAEs; any adverse event either reported for the first time or worsening of a pre-existing event after first dose of study drug until 31 days after the last dose of study drug) and serious TEAEs, including clinically significant changes in laboratory measures and vital signs. Lack of efficacy and failure of expected pharmacologicaction (e.g.‚ disease progression) were not reported as TEAEs. Additional details including timing of assessments are provided in the Supplemental Digital Content (https://links.lww.com/CCM/H210).
Statistical AnalysesThe primary endpoint was tested for each ruxolitinib group using a mixed-effects logistic regression model including treatment (ruxolitinib vs placebo) and ARDS severity (severe vs mild/moderate) as fixed effects and investigational site as a random intercept effect. Patients lost to follow-up before day 29 were not evaluable for the primary analysis but were included in a sensitivity analysis in which they were imputed as deaths. A sample size of 500 patients (pairwise comparison of 200 randomized to each ruxolitinib group and 100 to placebo) was initially planned to achieve approximately 83% power to detect a statistically significant effect of ruxolitinib with a nominal one-sided type I error of 1.44% (Dunnett’s procedure), assuming a mortality rate of 40% for ruxolitinib versus 60% for placebo based on limited mortality data emergent early in the pandemic (8,26). Owing to slow enrollment and an expected decline in U.S. cases with vaccine uptake, preliminary negative results from a separate trial of ruxolitinib in nonintubated hospitalized patients with COVID-19–associated cytokine storm (NCT04362137), and approval and commercial availability of another JAK1/JAK2 inhibitor (baricitinib) for this indication, the sponsor prematurely halted enrollment into RUXCOVID-DEVENT; final analyses were performed using all randomized patients (n = 211). The type I error level for the primary endpoint in this analysis was one-sided 2.5%; a fixed-sequence testing procedure was used to test the ruxolitinib 15 mg twice-daily group versus placebo first, and if significant, the 5 mg twice-daily group was tested. Use of one-sided type I error was based on the one-sided hypothesis that ruxolitinib added to standard of care would reduce 28-day mortality versus placebo plus standard of care. Two-sided CIs were presented for estimation of the mortality rate in both directions. Secondary efficacy variables were tested at the 0.05 level using a two-sided test; no type 1 error was allocated. Post hoc analyses of the primary endpoint for each ruxolitinib group in the U.S.-only population and pooled ruxolitinib treatment regimens in the full population were performed using similar methodology to the primary analysis.
The intention-to-treat population (all randomized patients) was used for analysis of summary demographics, baseline characteristics, patient disposition, and all efficacy outcomes unless otherwise noted. The safety population included all patients who received at least one dose of trial drug.
RESULTSBetween May 24, 2020, and December 15, 2020, 211 patients underwent randomization and were included in the intention-to-treat population (ruxolitinib 15 mg, n = 77; ruxolitinib 5 mg, n = 87; placebo, n = 47) (Fig. 1). All but two patients (both in the placebo group) received at least one dose of blinded study treatment and were included in the safety population. Among all patients, mean (sd) age was 63.4 (12.7) years (all ≥ 18 yr), 65% (137/211) were male, and 71% (149/211) were White (Table 1). Nine percent of patients (18/211) were enrolled in Russia and 91% (193/211) in the United States. Patient demographics and baseline clinical characteristics were generally balanced across groups, with a slightly higher proportion of men receiving placebo. Overall, 90% of patients (190/211) received prior or concomitant corticosteroids. Mean (sd) SOFA score at baseline was 9.4 (2.5), and baseline WHO nine-point ordinal scores were 6 and 7 for 48% of patients (101/211) and 52% of patients (110/211), respectively. ARDS severity at randomization was categorized as mild/moderate in 73% of patients (153/211) and as severe in 27% of patients (58/211). Comorbid conditions were present in most patients. Median (interquartile range) baseline IL-6 was 27.9 (11.3–114.1) pg/mL (eTable 2, https://links.lww.com/CCM/H210).
TABLE 1. - Patient Demographics and Baseline Clinical Characteristics of the Intention-to-Treat Population Characteristics Ruxolitinib 15 mg BID (n = 77) Ruxolitinib 5 mg BID (n = 87) Pooled Ruxolitinib (n = 164) Placebo (n = 47) Total (n = 211) Age, mean (sd), yr 63.6 (12.9) 63.6 (12.3) 63.6 (12.5) 62.5 (13.3) 63.4 (12.7) Male, n (%) 48 (62) 55 (63) 103 (63) 34 (72) 137 (65) Race, n (%) White 57 (74) 61 (70) 118 (72) 31 (66) 149 (71) Black 9 (12) 12 (14) 21 (13) 5 (11) 26 (12) Other/unknown 11 (14) 14 (16) 25 (15) 11 (23) 36 (17) Body mass index, mean (sd), kg/m2 34.8 (9.8) 33.5 (7.2) 34.1 (8.5) 33.7 (7.4) 34.0 (8.3) Country, n (%) United States 71 (92) 78 (90) 149 (91) 44 (94) 193 (91) Russia 6 (8) 9 (10) 15 (9) 3 (6) 18 (9) Predefined comorbidities,an (%) 66 (86) 76 (87) 142 (87) 36 (77) 178 (84) Hypertension 56 (73) 63 (72) 119 (73) 24 (51) 143 (68) Diabetes 40 (52) 43 (49) 83 (51) 23 (49) 106 (50) Chronic obstructive pulmonary disease 15 (19) 14 (16) 29 (18) 4 (9) 33 (16) Chronic kidney disease 7 (9) 14 (16) 21 (13) 7 (15) 28 (13) Chronic heart disease 10 (13) 10 (11) 20 (12) 6 (13) 26 (12) Asthma 4 (5) 15 (17) 19 (12) 4 (9) 23 (11) Sequential Organ Failure Assessment score, mean (sd) 9.2 (2.4) 9.6 (2.5) 9.4 (2.5) 9.4 (2.7) 9.4 (2.5) COVID-19 9-point ordinal scale, n (%)b 6 37 (48) 41 (47) 78 (48) 23 (49) 101 (48) 7 40 (52) 46 (53) 86 (52) 24 (51) 110 (52) ARDS severity at baseline, n (%) Pao 2/Fio 2 >100–300 mm Hg 59 (77) 66 (76) 125 (76) 33 (70) 158 (75) Pao 2/Fio 2 ≤100 mm Hg 18 (23) 20 (23) 38 (23) 14 (30) 52 (25) Missing 0 1 (1) 1 (1) 0 1 (<1) ARDS severity at randomization, n (%) Pao 2/Fio 2 >100–300 mm Hg 58 (75) 66 (76) 124 (76) 29 (62) 153 (73) Pao 2/Fio 2 ≤100 mm Hg 19 (25) 21 (24) 40 (24) 18 (38) 58 (27) Time from initial diagnosis to randomization, median (IQR), d 9.5 (5.0–15.0) 10.0 (6.0–14.0) 10.0 (5.0–15.0) 9.0 (4.0–15.0) 10.0 (5.0–15.0) Time from start of mechanical ventilation to randomization, median (IQR), hr 64.2 (37.9–104.5) 57.0 (32.3–144.4) 61.0 (34.6–120.3) 65.0 (34.8–168.0) 61.7 (34.7–121.4) ≤ 48, n (%) 29 (38) 35 (40) 64 (39) 17 (36) 81 (38) > 48, n (%) 48 (62) 52 (60) 100 (61) 30 (64) 130 (62) Prior or concomitant medications, n (%) Concomitant anticoagulant 72 (94) 82 (94) 154 (94) 42 (89) 196 (93) Corticosteroid 67 (87) 79 (91) 146 (89) 44 (94) 190 (90) Remdesivir 42 (55) 44 (51) 86 (52) 29 (62) 115 (55) Corticosteroid + remdesivir 37 (48) 42 (48) 79 (48) 28 (60) 107 (51) Convalescent plasma 11 (14) 16 (18) 27 (16) 10 (21) 37 (18) Prior biologic 3 (4) 8 (9) 11 (7) 0 11 (5)ARDS = acute respiratory distress syndrome‚ BID = twice a day‚ IQR = interquartile range.
aReported in > 10% of overall population.
bScale ranges from 0 to 8; score of 6 defined as intubation and mechanical ventilation and 7 as ventilation plus additional organ support (vasopressors, renal replacement therapy, or extracorporeal membrane oxygenation).
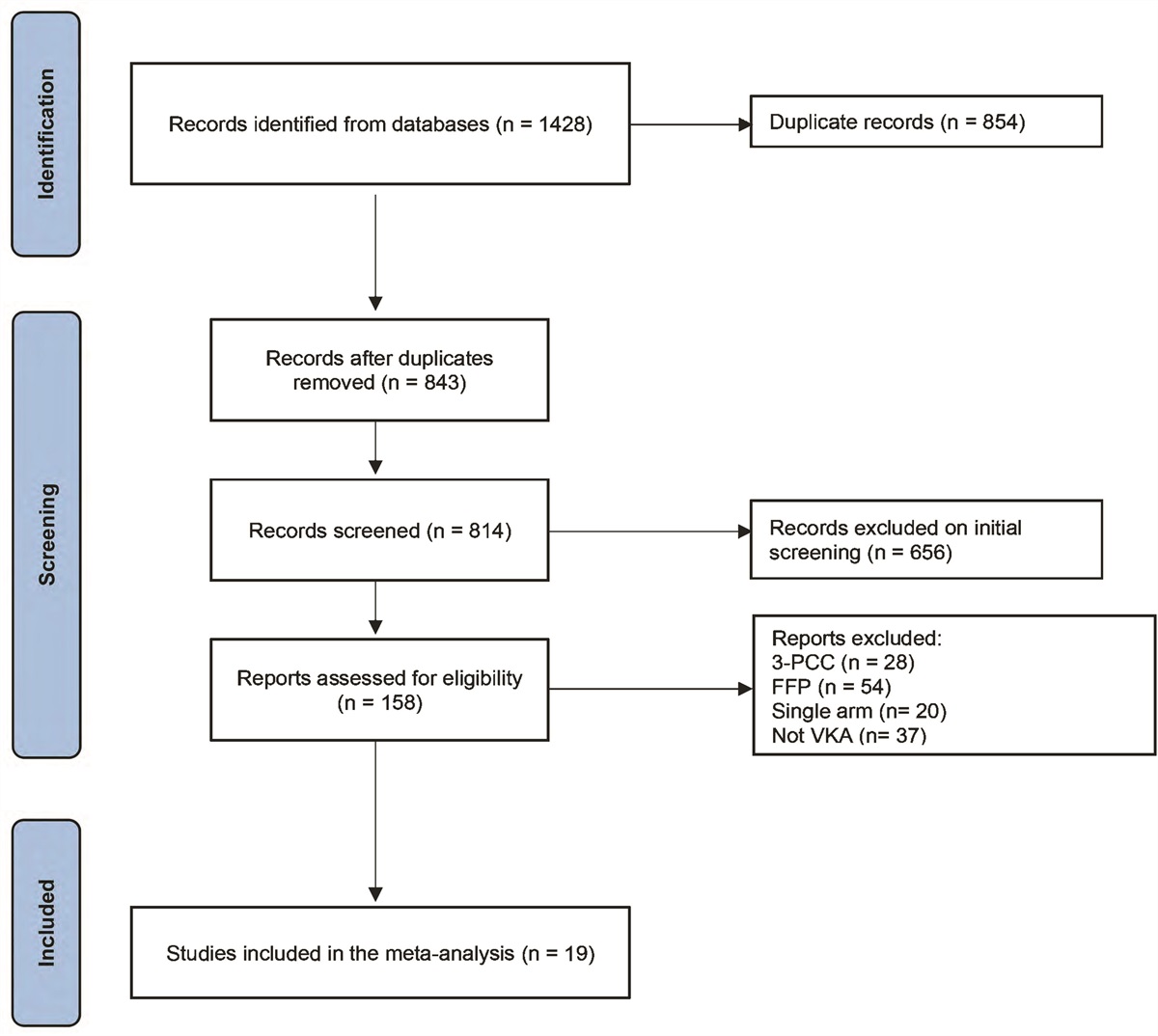
 Figure 1.:
Figure 1.: Trial profile. Efficacy analyses were conducted in the intention-to-treat population. *Patient numbers shown are for initial treatment; 73 patients entered the optional second 14-d treatment period (ruxolitinib 15 mg, n = 28; ruxolitinib 5 mg, n = 35; placebo, n = 10). †Reasons for physician decision included increasing creatinine levels (n = 1 [ruxolitinib 15 mg]), disease progression (n = 2 [ruxolitinib 15 mg, n = 1; placebo, n = 1]), and other reasons associated with worsening clinical status (n = 12 [ruxolitinib 15 mg, n = 4; ruxolitinib 5 mg, n = 4; placebo, n = 4]). ‡Patient no longer required treatment. ALT = alanine aminotransferase‚ AST = aspartate aminotransferase‚ CrCl = creatinine clearance‚ ECMO = extracorporeal membrane oxygenation‚ ULN = upper limit of normal.
Forty-five percent of patients (95/211) completed the entire 14-day protocol-defined ruxolitinib/placebo course (ruxolitinib 15 mg, 51% [39/77]; ruxolitinib 5 mg, 48% [42/87]; placebo, 30% [14/47]), and 73 patients entered the optional second 14-day treatment period (ruxolitinib 15 mg, n = 28; ruxolitinib 5 mg, n = 35; placebo, n = 10) (Fig. 1). The most common reasons for treatment discontinuation were death (ruxolitinib 15 mg, 29% [22/77]; ruxolitinib 5 mg, 32% [28/87]; placebo, 49% [23/47]), physician decision (ruxolitinib 15 mg, 8% [6/77]; ruxolitinib 5 mg, 5% [4/87]; placebo, 11% [5/47]), and AEs (ruxolitinib 15 mg, 9% [7/77]; ruxolitinib 5 mg, 6% [5/87]; placebo, 2% [1/47]).
The 28-day mortality rate was 51% (39/77; 95% CI, 39–62%) for ruxolitinib 15 mg and 53% (45/85; 95% CI, 42–64%) for ruxolitinib 5 mg versus 70% (33/47; 95% CI, 55–83%) for placebo (Table 2). The odds ratios (ORs) were 0.46 (95% CI, 0.201–1.028; one-sided p = 0.029) and 0.42 (95% CI, 0.171–1.023; one-sided p = 0.028) for 15 mg and 5 mg, respectively, versus placebo. Results of the sensitivity analysis are shown in eTable 3 (https://links.lww.com/CCM/H210). Subgroup analyses of 28-day mortality demonstrated consistent trends across patient demographic and clinical characteristic subgroups (eFig. 2, https://links.lww.com/CCM/H210). Eight of 11 patients (all ruxolitinib-treated) who received prior biologics (all anti–IL-6 antibodies) died by day 29. In a post hoc analysis of the primary endpoint evaluating the U.S. population only (n = 191), the 28-day mortality rate was 46.5% (33/71; 95% CI, 34.5%–58.7%) for the ruxolitinib 15 mg treatment group and 47.4% (36/76; 95% CI, 35.8%–59.2%) for ruxolitinib 5 mg versus 68.2% (30/44; 95% CI, 52.4%–81.4%) for placebo (eTable 4, https://links.lww.com/CCM/H210). The ORs were 0.43 (95% CI, 0.188–0.974; one-sided p = 0.022) and 0.39 (95% CI, 0.157–0.948; one-sided p = 0.019) for ruxolitinib 15 mg and 5 mg, respectively, versus placebo. Further, post hoc analysis of the primary outcome pooling the 5 mg and 15 mg ruxolitinib doses for the overall population demonstrated a 28-day mortality rate of 51.9% (84/162; 95% CI, 43.9%–59.8%) with ruxolitinib versus 70.2% (33/47; 95% CI, 55.1%–82.7%) for placebo (OR, 0.47 [95% CI, 0.219–0.996]; one-sided p = 0.024) (eTable 4, https://links.lww.com/CCM/H210).
TABLE 2. - Primary and Secondary Efficacy Outcomes (Intention-to-Treat Population) Outcome Ruxolitinib 15 mg BID (n = 77) Ruxolitinib 5 mg BID (n = 87) Placebo (n = 47) Primary outcome 28-d mortality Death due to any cause prior to or on Day 29, n/N (%) 39/77 (51) 45/85a (53) 33/47 (70) 95% CI for 28-d mortality rate 39.0–62.2 41.8–63.9 55.1–82.7 Odds ratio (95% CI) for ruxolitinib vs placebo 0.46 (0.201–1.028) 0.42 (0.171–1.203) NA One-sided p 0.029 0.028 NA Secondary outcomes In-hospital outcomes Mean (sd) P b Mean (sd) P b Mean (sd) Ventilator-free days 6.3 (9.0) 0.015 4.9 (8.4) 0.131 3.0 (7.2) ICU-free days 4.8 (7.7) 0.017 4.0 (7.5) 0.107 2.5 (6.4) Vasopressor-free days 9.0 (11.7) 0.014 7.5 (10.9) 0.051 4.5 (9.3) Hospital-free days 2.1 (4.9) 0.190 2.4 (5.3) 0.155 1.4 (4.0) Oxygen-free days 2.7 (6.1) 0.201 3.0 (6.7) 0.212 1.5 (4.7) COVID-19 nine-point ordinal scale score Time to any improvement, median (95% CI) 9.0 (7.0–14.0) 13.0 (8.0–19.0) 11.0 (8.0–not evaluable) Day 15, n (%) 0–2 (uninfected/ambulatory) 4 (5) 5 (6) 2 (4) 3–5 (hospitalized, not intubated) 18 (23) 13 (15) 3 (6) 6–7 (intubated) 31 (40) 42 (48) 13 (28) 8 (dead) 24 (31) 25 (29) 29 (62) ≥ 1-point improvement from baseline, n/N (%) 32/77 (42) 28/85 (33) 10/47 (21) Odds ratio (95% CI) for ruxolitinib vs placebo 2.54 (1.067–6.034) 1.72 (0.732–4.041) NA P c 0.0351 0.213 NA ≥ 2-point improvement from baseline, n/N (%) 17/77 (22) 13/85 (15) 5/47 (11) Odds ratio (95% CI) for ruxolitinib vs placebo 2.15 (0.724–6.358) 1.32 (0.431–4.036) NA P c 0.169 0.628 NA Change from day 1 to day 15, mean (sd) –0.4 (1.8) –0.2 (1.7) 0.6 (1.7) Day 29, n (%) 0–2 (uninfected/ambulatory) 15 (19) 18 (21) 6 (13) 3–5 (hospitalized, not intubated) 10 (13) 6 (7) 2 (4) 6–7 (intubated) 10 (13) 14 (16) 6 (13) 8 (dead) 39 (51) 45 (52) 33 (70) ≥ 1-point improvement from baseline, n/N (%) 32/74 (43) 27/83 (33) 8/47 (17) Odds ratio (95% CI) for ruxolitinib vs placebo 3.48 (1.406–8.624) 2.28 (0.899–5.789) NA P c 0.0070 0.0824 NA ≥ 2-point improvement from baseline, n/N (%) 22/74 (30) 21/83 (25) 8/47 (17) Odds ratio (95% CI) for ruxolitinib vs placebo 1.89 (0.751–4.759) 1.58 (0.616–4.035) NA P c 0.177 0.342 NA Change from day 1 to day 29, mean (sd) –0.5 (2.5) –0.4 (2.6) 0.4 (2.2)BID = twice a day, NA = not applicable.
aTwo patients in the ruxolitinib 5 mg BID group were not evaluable for analysis of the primary endpoint (withdrawn consent).
bPer Kruskal-Wallis test; tested (vs placebo) at the 0.05 level using a two-sided test with no type 1 error allocated.
cPer Wald test from a logistic regression model that included treatment group and acute respiratory distress syndrome severity as fixed effects and investigational site as a random effect; tested (vs placebo) at the 0.05 level using a two-sided test with no type 1 error allocated.
Table 2 shows secondary efficacy outcomes. Patients receiving 15 mg ruxolitinib showed numerical improvements for in-hospital outcomes, including number of ventilator-free, ICU-free, and vasopressor-free days, versus placebo. Hospitalization outcomes for the U.S. population only are presented in eTable 5 (https://links.lww.com/CCM/H210). Patients treated with ruxolitinib showed a numerical improvement from baseline on the nine-point ordinal scale at days 15 and 29 versus placebo. Ordinal scale scores by trial day (days 1–29) for each treatment group are shown in Figure 2. SOFA scores tended to increase (indicating clinical decline) more gradually with ruxolitinib compared with placebo (eFigs. 3 and 4, https://links.lww.com/CCM/H210).
 Figure 2.:
Figure 2.: COVID-19 nine-point ordinal scale score by day. A, Ruxolitinib 15 mg twice a day (BID), B, ruxolitinib 5 mg BID, or C, placebo (intention-to-treat population). LTFU = lost to follow-up.
Overall rates of TEAEs were similar across trial groups (Table 3). The most common TEAEs among patients treated with ruxolitinib (15 mg/5 mg; vs placebo) were anemia (17% [13/77]/24% [21/87] vs 22% [10/45]) and pneumonia (17% [13/77]/16% [14/87] vs 20% [9/45]). The most common grade 3 or higher TEAEs with ruxolitinib versus placebo were pneumonia (16% [12/77]/13% [11/87] vs 18% [8/45]) and anemia (12% [9/77]/14% [12/87] vs 18% [8/45]) (eTable 6, https://links.lww.com/CCM
留言 (0)