
記住我


The glutaredoxin-mediated reduction of protein mixed disulfides to protein thiols is critical in the brain where glutathionylation of protein is more prominent. In the brain, under conditions of oxidative stress such as that seen during reperfusion following ischemia or in animal models of MPTP, there is a predominant formation of Pr-SS-Pr. During recovery following these insults, Pr-SSG is reduced back to Pr-SH by Grx1, thus restoring protein function.
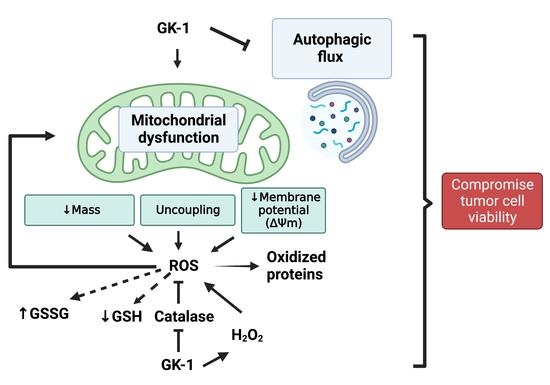
Glutaredoxin was first purified by Mieyal et al. from human erythrocytes as thioltransferase specific for GSH containing mixed disulfides [41] and cloned in Escherichia coli [42]. In rat brains, a constitutive expression of glutaredoxin exhibits regional and cellular variability in its localization. Glutaredoxin activity is detected higher in the hippocampus where they could recover more rapidly from oxidative damage compared to other regions with less activity in the cerebellum and striatum [43]. A similar study carried out using human autopsy brain samples detected higher glutaredoxin activity in the hippocampus and cerebellum. Glutaredoxins are localized predominantly in neurons in the cerebral cortex and hippocampus, purkinje and granule cell layers of the cerebellum, and granular cell layers of the dentate gyrus in the human brain [44]. 1.3. Glutaredoxin and Mitochondrial DysfunctionMaintaining protein thiol homeostasis is important for mitochondrial functions. Loss of GSH and formation of Pr-SSG in the brain contributes to various dysfunctions including loss of activity of mitochondrial enzymes, structural damage to mitochondrial membranes and deregulation of cell survival pathways. Mitochondria are prone to oxidative damage as ROS generated during oxidative phosphorylation can in turn releases more reactive oxygen species that can damage the macromolecules essential for its normal function. The percentage of total cell GSH present in mitochondria is 10–15% [45].The response of brain mitochondria to oxidative stress is very different from the liver mitochondria. In the brain, the GSH lost during oxidative stress is recovered as Pr-SSG and only less than 5% of GSH is recovered as GSSG [46]. However, in the liver, most of the GSH lost is recovered as GSSG, and a reduced state of protein thiols are maintained. The excess GSSG formed is effluxed out of the cell to prevent oxidative modification of proteins [47].Mitochondrial enzymes are particularly vulnerable to oxidative damage, complex Ⅰ being the most affected as it increases the oxidation of the GSH pool. Protein thiol groups on 75 and 51 KDa subunits of complex Ⅰ undergo glutathionylation and forms mixed disulfide with GSH [48] (Figure 2). Thiol modifications on complex Ⅰ inhibit its activity and increase ROS production that can be reversed by either Grx1 or the disulfide reducing agent dithiothreitol [49]. Downregulation of Grx1 in the brain regions of Swiss albino mice results in a significant loss of complex Ⅰ activity signifying the role of glutaredoxins in mitochondrial function [50]. ROS-mediated inhibition of activity is specific to complex Ⅰ while other complexes in the electron transport chain remain unaffected.Glutaredoxin 2 (Grx2) was first identified and cloned by Holmgren et al., which is 34% identical to cytosolic Grx1 [51]. It has three isoforms: Grx2a is localized to mitochondria, and Grx2b and Grx2c are both localized to the nucleus and cytosol [52]. Grx2a, a mitochondrial isoform of glutaredoxin regulates the glutathionylation of mitochondrial enzymes. Constitutive expression of Grx2 is observed in mouse and human brains and is localized to neurons and glia cells including the neurons of substantia nigra. Grx2 expression is transiently upregulated in MPTP-treated mice and a partial loss in complex Ⅰ activity due to Grx2 downregulation [53]. Theoverexpression of Grx2 prevents apoptosis by inhibiting cytochrome c release and caspase activation [54] and could ameliorate the toxic effect of MPP+ in mitochondria [53].Further, overexpression of Grx2 attenuatesthe mutant superoxide dismutase (SOD1) mediated degeneration of motor neurons. MutSOD1 accumulates in the mitochondria of motor neurons, and impairs respiratory complexes and ATP production. Grx2 is required for normal mitochondrial function and viability, whereas overexpression of Grx1 is shown to be beneficial in the cytosol and does not preserve mitochondrial dynamics or apoptosis induced by mutSOD1 [55].Increased Pr-SSG formation due to loss of Grx1 activity in the cytosol is known to impair mitochondrial dynamics in the brain. Pr-SSG formed by the oxidation of sulfhydryl groups of cysteine increases the permeability of the inner mitochondrial membrane and opens upa mitochondrial transition pore, resulting in the uncoupling of oxidative phosphorylation and ATP hydrolysis. In addition, the oxidation of voltage-dependent anion channel (VDAC), a mitochondrial outer membrane protein results in the loss of mitochondrial membrane potential (MMP Figure 4). The redox state of vicinal thiol groups in VDAC plays a critical role in the tuning of the voltage sensor of the transition pore and increases its permeability upon oxidation [56]. Moreover, VDAC is present in the outer membrane of mitochondria and is exposed to cytosolic oxidative stress as well. shRNA mediated downregulation of Grx1 results in the oxidation of VDAC but not adenosine nucleotide translocase (ANT), an inner mitochondrial membrane protein. Exposure of Neuro2A cells to β-N-oxalyl amino-L-alanine (L-BOAA), an excitatory amino acid implicated in neurolathyrism also leads to MMP loss, which is alleviated by overexpression of Grx1 [57]. MMP loss results in cristae unfolding in mitochondria facilitating the release of apoptotic factor cytochrome c leading to cell death [58]. Thus, Grx1 functions to maintain mitochondrial integrity during oxidative stress and the downregulation of Grx1 results in mitochondrial dysfunction through oxidative modification of the thiol group present in the outer membrane protein VDAC.Overall, glutaredoxin acts as a potential neuroprotective mediator to maintain mitochondrial function during oxidative stress, excitatory amino acid toxicity and MPTP mediated neurotoxicity. The role of glutaredoxins will be discussed under each of the disease conditions that follow.
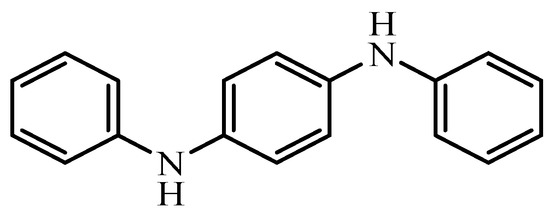
1.6. Parkinson’s DiseasePD is a movement disorder, characterized by the presence of Lewy bodies as intracellular aggregates of α-synuclein and loss of dopaminegic neurons from substantia nigra pars compacta regions (SNpc) [67]. Although the cause of PD is multifactorial, oxidative stress plays a major role in the degeneration of dopaminergic neurons in PD [26]. Several genes identified including parkin, α-synuclein, DJ1, PINK1, and LRRK2, whose mutations are responsible for familial PD, map on to disruption of the ROS scavenging pathway and mitochondrial dysfunctions [68,69,70,71] (Figure 5).Dopaminergic neurons produce dopamine (DA), a neurotransmitter that modulates synaptic transmission to control motor activity. Dopaminergic neurons are rich in neuromelanin and iron content [73,74]. Both ferrous (Fe2+) and ferric states (Fe3+) of iron are present in Lewy bodies and iron content is higher in the SNpc regions of the PD brain. When an excess amount of redox-active iron is present in a cell, it generates more ROS, such as superoxides and hydroxyl radicals or lipid peroxides via fenton reaction [75]. The fenton reaction is a chemical reaction between ferrous iron and hydrogen peroxides to form ferric iron and highly reactive hydroxyl radicals (HO•) (Equation (1)). Ferric iron formed reacts with superoxides generated in mitochondria to reduce back to ferrous state (Equation (2)). Hydroxyl radicals are thus formed and react with lipids to form lipid radicals or lipid peroxyl radicals called malondialdehyde (Equation (3)).Fe2++H2O2→Fe3++HO−+HO•
(1)
Fe3++O2•−→Fe2++O2
(2)
HO•+LH(lipids)→L•(Lipid radicals)+H2O
(3)
Iron is mostly associated with proteins in the cell and readily incorporated into mitochondria. Iron-containing enzymes in mitochondria also generates a substantial amount of ROS through a biological process [76,77]. Additionally, DA can be oxidized by iron to produce DA-quinones that can react with the sulfhydryl group of cysteine to modify proteins causing damage to cells and membranes, thus leading to cell death [78,79].Several animal models are utilized to study different aspects of PD pathology, which include genetic and neurotoxin-induced models. Genetic models, linked to monogenic PD, include a mutation in genes SNCA, LRRK2, UCH-L1, PRKN, and PINK1, and exhibit dopaminergic cell loss, motor deficit and mostly formation of Lewy bodies. Animal models utilizing neurotoxins such as MPTP, rotenone, diamide, and herbicide paraquat, induce loss of dopaminergic neurons and elicit motor symptoms, but mostly lack α-synuclein aggregation seen as Lewy body formation [80].MPTP is the most common neurotoxin used to induce parkinsonism to identify early events in PD pathology [81,82]. MPTP increases the number of reactive oxygen species and lipid peroxidation product and is accompanied by a loss in GSH in both the in vitro and the in vivo mouse model. In vivo administration of MPTP results in the lowering of GSH along with increased ROS in midbrain and striatum [83]. MPP+, an active metabolite of MPTP (Figure 6) administration inhibits mitochondrial complex Ⅰ activity [84] and is confined to dopaminergic cells which, in part, explains their susceptibility towards oxidative damage in PD [85].Mitochondrial dysfunction is implicated in PD neurodegeneration [86,87]. Several familial PD genes parkin, PINK1 and DJ1 are linked to mitochondrial pathways (Figure 5). Grx1 is essential for maintaining mitochondrial functions in brain treated with MPTP. They are also involved in the recovery of complex Ⅰ activity after MPTP treatment, which is shown to be impaired with the down regulated Grx1 [50].In addition to mitochondrial dysfunction, dephosphorylation of Akt1 is seen in MPTP treated animal models. Akt1, a serine/threonine kinase critical for cell survival, undergoes oxidative modification and its impaired activity is observed in PD. Phosphorylated Akt1 mediates cell survival by inhibiting proapoptotic proteins, Bad, caspase and transcription factor FOXO. The oxidation of cysteine residue in Akt1 leads to dephosphorylation, loss of 40% reduced form and inhibition of kinase activity in MPTP treated mice. When cysteine residues at 296 and 310 of Akt1 are oxidized, it increases its association with protein phosphatase2A leading to dephosphorylation [88]. Exposure of cadmium, a pan inhibitor of protein thiol sulfide oxidoreductase or the downregulation of Grx1 in the Neuro2A cell line results in dephosphorylation of Akt1 and decreased Akt1 protein levels. Mutation of critical cysteines at 296 and 310 of Akt1 to serine prevents oxidative modification and increases the cell survival effect of Akt1. MPP+ mediated loss in Akt levels is ameliorated by overexpression of Grx1. Therefore, maintaining the thiol status in Akt1 protects it from proteasomal degradation and would be beneficial for cell survival during oxidative damage [89].Indeed, if Grx1 downregulation causes a similar effect in protein thiol modification as seen with the MPTP model, then a thiol oxidizing agent such as diamide, or diazenedicarboxylic acid bis (N, N-dimethylamide) (Figure 6), would also cause neurodegeneration. One molar equivalent diamide reversibly oxidizes GSH to form a molar equivalent of thiol-disulfide and promotes the conversion of NADPH to NADP+ in less than a minute. Diamide-induced GSH loss persists for a substantial period, unlike transient ischemia and MPTP models which restore thiol homeostasis after an initial insult. Single focal delivery of diamide to substantia nigra targets protein thiols and triggers Parkinsonism phenotypes, such as α-synuclein aggregation, locomotor deficit, and DA neurodegeneration in mice. Disruption of protein thiol homeostasis by diamide triggers neurodegenerative cascade in SNpc neurons and activates the ASK1-p38 MAPK death signaling pathway [90]. Activation of the ASK1-p38 MAPK pathway is a common feature across different PD models. Activated ASK1 mediates the downstream MAPK signaling via p38 MAPK phosphorylation. Downregulating ASK-1 attenuates DA neurodegeneration, indicating its role in neurodegeneration in SN regions [91]. Altogether, maintaining protein thiol homeostasis is important for DA neuron survival as a single administration of diamide causes sustained changes in biochemical and behavioral parameters.Furthermore, the selective downregulation of Grx1 in SNpc shows a loss of TH positive neurons and the development of motor deficits. A recent study concludes that the knockdown of Grx1 in substantia nigra could selectively degenerate SNpc DA neurons without affecting them in the VTA region. Moreover, significant downregulation of Grx1 mRNA was also observed in the SNpc region of human PD autopsy tissue [92].It is very well known that typical neuroleptic such as haloperidol, while being a very effective anti-psychotic drug, is known to cause tardive dyskinesia in a subset of patients. Symptoms of dyskinesia overlapping with PD is a very big concern that led to the development of atypical anti-psychotic drugs such as clozapine, quetiapine, etc [93]. Haloperidol induces oxidative stress by increasing the turnover rate of dopamine and shows a similar effect as seen in MPTP in terms of protein thiol oxidation. Acute [94] and chronic [95] administration of haloperidol in rats result in the loss of GSH along with an increase in malondialdehyde, a lipid peroxidation product. A single dose of haloperidol also causes selective inhibition of mitochondrial complex Ⅰ due to the oxidation of essential thiol groups that can be reversed by thiol antioxidant α-lipoic acid [96,97].Paraquat, a herbicide with structural similarity to MPP+, also induces dopaminegic neurodegeneration and mitochondrial dysfunction via oxidative stress, mimicking features of pathological PD [98] (Figure 6). It selectively targets neurons in the SNpc sparing VTA region similar to the observations seen in the PD brain [99,100]. Paraquat generates ROS by redox cycling reaction, in which the herbicide undergoes one-electron reduction to form a paraquat ion followed by an electron transfer to molecular oxygen leading to the formation of superoxides [101] (Figure 6). Paraquat-induced neuronal death is mediated by the phosphorylation of c-Jun N-terminal kinase (JNK) and c-Jun where it activates caspase-3 and apoptosis [102]. Paraquat may inhibit mitochondrial functions through an oxidative mechanism but not through direct inhibition of complex Ⅰ, as in the case of MPTP [98]. Studies conducted to determine the role of Grx1 and protein glutathionylation in paraquat induced nigrostriatal degeneration show a similar effect as seen with MPTP [103]. Indeed, the common features associated with MPTP, diamide, haloperidol, and paraquat, emphasize the selective vulnerability of dopaminergic neurons to oxidative stress and the importance of protein thiol homeostasis.Several studies on the effect of antioxidant, α-lipoic acid (ALA) across different PD models with a motor deficit and neuroinflammation found promising results in alleviating the symptoms of PD. ALA regulates ferroptosis, a programmed cell death that depends on iron and increased lipid peroxides via activating PI3K/Akt/Nrf2 pathway [104]. ALA reduces the expression levels of inflammatory mediators such as NF-κB, TNF-α, and iNOS, in substantia nigra and improves motor performance by increasing locomotor activity [104]. 1.7. Alzheimer’s Disease (AD)Alzheimer’s disease is an age-related progressive form of dementia, characterized by the presence of extracellular amyloid-β deposition, neurofibrillary tangle formations, loss of synapses and neurodegeneration. Multiple pathways contribute to AD pathogenesis and oxidative stress is one of the early events [105,106]. Aβ-42 (Aβ-42) which has a causative role in AD itself produces ROS. Amyloid-β in aqueous solution fragments and generates free radical peptides identified via electron paramagnetic resonance spin trapping. Aβ fragments produce reactive oxygen species and inactivate glutamine synthetase, which is essential for glutamate metabolism [107,108].Synaptic dysfunction including loss of dendritic spines is one of the major consequences of AD [109] which affects activity dependent protein translation and structural plasticity. Protein translation at the synapse is critical for its maintenance and plasticity. The synthesis of new proteins in response to neuronal stimulation is required for synaptogenesis. The Akt1-mTOR signalling pathway mediates most of the protein translation occurring at the cell and is critical for activity-dependent protein translation. Thiol oxidation of Akt1 due to oxidative stress contributes to synaptic dysfunction in APP/PS1 mice. The oxidation of cysteine groups in Akt1 increases its association with PP2A, causing dephosphorylation and inhibiting its kinase activity. The subsequent loss of Akt1-mTOR signaling affects activity-dependent protein translation at the synapse [110].Regulating actin dynamics is critical to synaptic functions including synaptic plasticity, memory and learning. The G-actin/F-actin equilibrium is maintained by several actin binding proteins, mainly Cofilin [111]. Cofilin undergoes oxidation in stressed neurons to form dimers and induces the formation of cofilin-actin rods in neurites. Rod formation causes synaptic dysfunctions by disrupting actin dynamics, sequestering dephosphorylated cofilin and increasing mitochondrial membrane potential loss. Cofilin signaling pathways are implicated in synaptic deficits associated with AD. In addition, cofilin-actin rods are observed in cognitively impaired human AD brains [112].The increased S-glutathionylationin proteins has been identified [113] along with a reduction in thiol oxidoreductase levels in the AD brain compared with age matched controls [114]. Actin, similar to many other proteins, is targets for S-glutathionylation [115,116]. Glutathionylated actin at Cys-374 residue decreases its polymerization rate [117] (Figure 7). A recent study looked at the role of amyloid-β-induced oxidative stress in glutathionylation of actin, showing F-actin loss in the AD mouse model. A reduced form of F-actin is significantly decreased along with an increase in F-actin and G-actin glutathionylation in APP/PS1 mice. Grx1 plays a critical role in maintaining the thiol status of the F-actin cytoskeleton in dendritic spines. The overexpression of Grx1 in the APP/PS1 hippocampus reverses the contextual fear conditioning (cFC) recall deficit, restoring F-actin levels and decreasing ROS levels. Spine morphology and F-actin nanoassembly are restored in primary cortical cultures overexpressed with Grx1. Ameliorating the levels of Grx1 in AD brains could potentially help in maintaining synaptic plasticity and memory deficit [118].
留言 (0)