
記住我
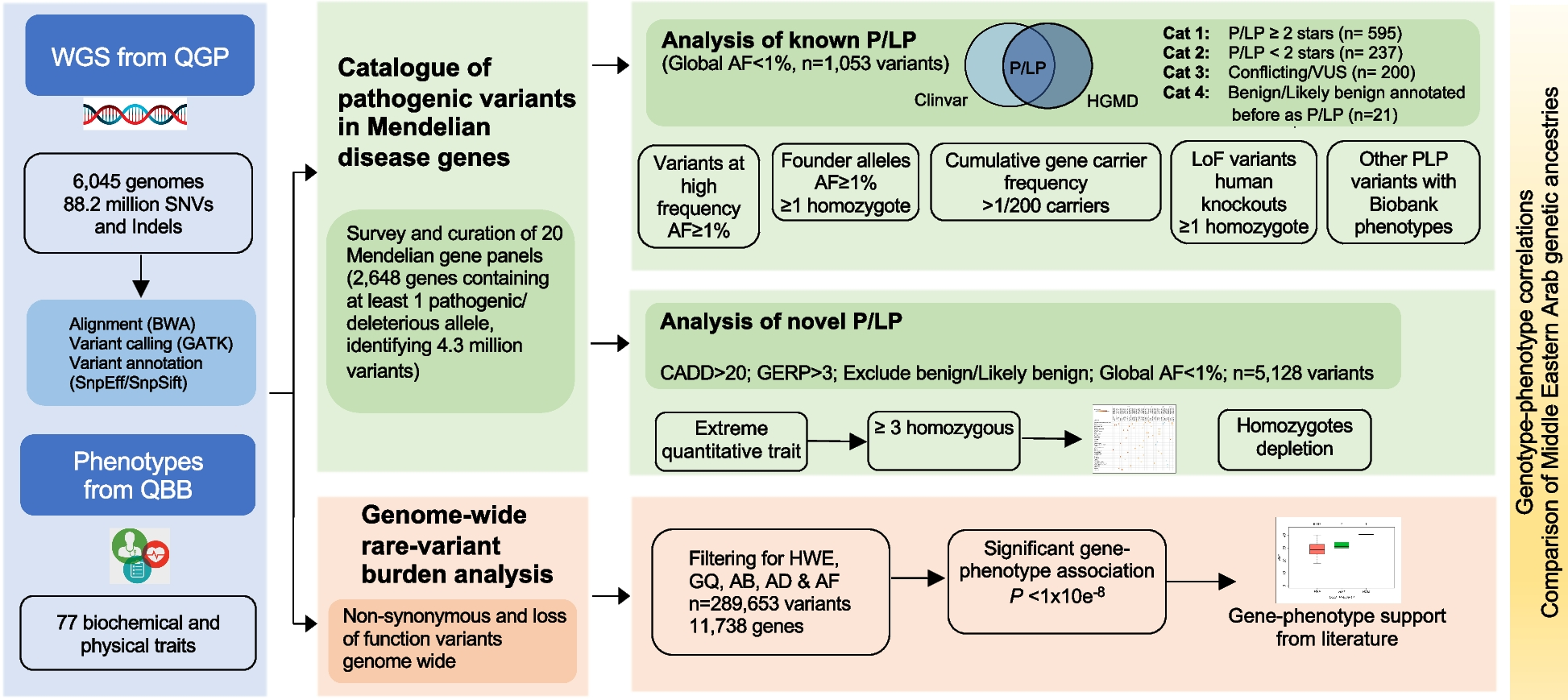
Trans-eQTLs are especially difficult to identify in cohorts of limited sample sizes. To overcome this challenge, we worked with a large number of samples (N=1387, which includes the PsychENCODE brain resource, CMC, and GTEx brain samples), thereby more readily enabling us to identify candidate trans-eQTLs. We investigated the extent to which the resulting candidate trans-eQTLs might reveal potential mechanisms of distal regulatory linkages across chromosomes. We tested associations between 12,245 highly expressed genes and autosomal variants on a genome-wide scale (see Methods for processing and filtering criteria). We used the same covariates as those used for identifying cis-eQTLs in our previously published work [10]. We removed genes with poor mappability and variants located in repetitive regions. Furthermore, to minimize false positives, we filtered out candidate trans-eQTLs between pairs of genomic loci with evidence of RNA-sequencing (RNA-seq) read cross-mapping [30]. We calculated false discovery rate (FDR) values from the linkage-disequilibrium (LD)-pruned list of candidate trans-eQTLs to determine significance. We employed the following three ways of performing multiple test correction to call trans-eQTLs, and we provide the lists called with each of these methods as Additional files.
1)Pooled genome-level FDR correction method per GTEx 2017 [17]. At an FDR threshold of 0.25, we detected 77,156 candidate trans-eSNPs from ~5.3M total SNPs tested in locations ≥5 Mb from the gene transcription start site (TSS), comprising 17,899 independent SNPs after LD pruning (Fig. 1A). We term this list (at FDR<0.25) “candidate trans-eQTLs” and provide it in Additional file 1. Separately, we compiled alternate lists at different confidence thresholds. In particular, we identified 7642 candidate trans-eQTLs involving 580 eGenes at an FDR threshold of 0.05; this list is available in Additional file 2.
2)Hierarchical gene-level FDR correction per GTEx 2017 [17]. This method generates an eGene list. We provide this list (at a gene-level FDR<0.25) in Additional file 3.
3)Permutation-based hierarchical gene-level FDR per GTEx 2020 [9]. We used this hierarchical multiple test correction via a permutation-based scheme and provide the list (at FDR<0.25) in Additional file 4.
Fig. 1
Characterization of candidate trans-eQTLs. A Genetic map for candidate trans-eQTLs. B Comparisons of the effect sizes between cis-eQTLs and candidate trans-eQTLs. C Frequencies with which SNPs are shared among various combinations of the 3 QTL types (cis-eQTLs, candidate trans-eQTLs, and cQTLs). D Enrichment statistics and variant proportions for the frequencies with which cis-eQTLs, candidate trans-eQTLs, and cQTLs lie within various types of genomic elements
We performed an internal replication validation for the candidate trans-eQTLs by splitting the samples randomly to two folds with no overlapping samples. Two sets of candidate trans-eQTLs were then called after this splitting: one set was called using only the samples in fold 1, and the other set was called using only the samples in fold 2 (i.e., the trans-eQTLs were calculated within each fold separately). We then quantified the similarities between these 2 sets of candidate trans-eQTLs by measuring the overlap of trans-eQTLs from each of the two folds. We found that the overlap of the trans-eQTLs generated by these two folds is around 80% at an FDR threshold of 0.05.
Relative to previously published studies, we identified substantially more candidate trans-eQTLs and associated eGenes in the human brain by leveraging integrated data resources. We used the candidate trans-eQTL list with FDR<0.25 for the analyses discussed in this study (i.e., those provided in Additional file 1).
We next characterized the genomic features of candidate trans-eQTLs and compared them with other types of QTLs in order to investigate associations between genomic elements and QTLs (Fig. 1B–D). In agreement with previous findings, we found that the magnitude of the effect sizes associated with cis-eQTLs are larger than that those of candidate trans-eQTLs (Fig. 1B). Candidate trans-eQTLs overlapped more frequently with cis-eQTLs than with chromatin QTLs (cQTLs) [10], which is also largely expected. The low overlap of cQTLs and candidate trans-eQTLs may be a consequence of the overall low number of cQTLs. We found 20,175 eSNPs that are shared between cis- and candidate trans-eQTLs, whereas only 61 SNPs were shared between candidate trans-eQTLs and cQTLs. As shown in Fig. 1C, we found 25 SNPs that are shared among all three QTL types (i.e., cis-eQTLs, candidate trans-eQTLs, and cQTLs). With respect to genomic elements, we found that candidate trans-eQTLs tend to exhibit lower enrichment in most elements relative to cis-eQTLs and cQTLs; however, candidate trans-eQTLs are most enriched within exons (Fig. 1D). The pattern of variant proportion on different genomic regions for candidate trans-eQTLs is similar to that for cis-eQTLs but not cQTLs. We also calculated the enrichment of the QTLs in previously published brain enhancer lists [31] and found the enrichment patterns to be similar across these enhancers (Additional file 6: Fig. S6A).
Potential mechanisms of candidate trans-eQTLsIn one potential mechanism, specific variants exert trans-regulatory effects by influencing a nearby regulatory factor (such as a TF), which in turn regulates distal genes in a trans fashion [3]. In this scenario, trans-eQTLs may also act as cis-eQTLs for nearby genes that have regulatory impacts on trans-eGenes (Fig. 2A). Indeed, we found that 19.33% of LD-pruned candidate trans-eQTLs display cis-eQTL signals (Fig. 2D). Because simple genomic coordinate-level overlaps between cis- and candidate trans-eQTLs may detect spurious associations due to LD, we performed a colocalization assay to identify cis- and candidate trans-eQTL pairs that harbor shared causal variants. In total, we detected 1688 candidate trans-eQTLs (48.79% of the candidate trans-eQTLs that overlap with cis-eQTLs) that have shared causal variants with cis-eQTLs. We further interrogated the potential causal effects of cis-eQTLs on trans-eQTL associations via mediation analysis (Fig. 2B, Additional file 5). As part of this analysis, we found that 62.13% of the candidate trans-eQTLs that colocalize with cis-eQTLs can be explained by cis-mediators (p<0.05). Among the candidate trans-eQTLs that can be explained by cis-mediators, there is roughly an even split of cis-mediators with positive and negative mediation effects, suggesting that trans-regulators can either be activators or repressors with roughly equal probability (Fig. 2B). As generally expected, we also observed that larger mediation coefficients tend to have greater statistical significance. We found that 85% of trans-eGenes are also cis-eGenes, indicating that gene expression is regulated by both cis- and trans-acting variants. Roughly 20% of trans-eGenes had cis-eGenes as mediators (Fig. 2D).
Fig. 2
Cis-mediation and inter-chromosomal interactions explain candidate trans-eQTL associations. A A candidate trans-eQTL may overlap with a cis-eQTL that regulates a proximal gene (cis-eGene), which may in turn regulate a distal gene (trans-eGene). B Roughly 62% of candidate trans-eQTLs colocalized with cis-eQTLs exhibit mediation effects. The number of candidate trans-eQTLs with positive mediation effect sizes is almost the same as those that are negative. C Cis- and trans-eGene pairs with evidence of cis-mediation display significant co-expression compared to both random pairs and those pairs that exhibit colocalization but not mediation. D Relative abundances of candidate trans-eQTLs that overlap with cis-eQTLs (left), colocalize with cis-eQTLs (middle), and exhibit mediation effects (right). Roughly 85% of trans-eGenes are also cis-eGenes, and ~17% of trans-eGenes have cis-mediators. E. Biological pathways associated with cis-eGenes that mediate candidate trans-eQTL associations are enriched for metabolic processes and transcriptional regulation. F Candidate trans-eQTL and eGene pairs tend to exhibit inter-chromosomal interactions more frequently than do random pairs. ACME, average causal mediation effects
By definition, because cis-mediation implies that variants influence trans-eGene expression via cis-eGenes, we hypothesized that cis- and trans-eGene pairs with evidence of cis-mediation are co-regulated. We evaluated this hypothesis by first grouping cis- and trans-eGene pairs into those that exhibit evidence of cis-mediation (which we term “mediation pairs”) and those with evidence of colocalization but not mediation (which we term “colocalization pairs”). When comparing these groups, we found that mediation pairs showed greater expression correlation than colocalization pairs or expression-level matched random pairs (Fig. 2C; see the “Methods” section), thereby providing additional evidence for cis-mediation. We also observed that trans-eSNPs are enriched on exons (Fig. 1D). Therefore, one possible mediating mechanism may indeed be non-synonymous coding variation in the mediating genes (Additional file 6: Fig. S6B).
We next investigated the properties of cis-eGenes that were found to mediate trans-regulatory effects. In addition to being enriched for transcriptional regulators (e.g., members of TF complexes), cis-eGenes were also enriched for other biological processes (e.g., metabolic processes, Fig. 2E). These results indicate that variant effects on distal gene regulation are not solely dependent on TF activity. Instead, variants that are associated with metabolism may exert broad systems-level effects on cellular function, which can then lead to changes in distal gene expression.
Another potential explanation for trans-eQTLs may lie in inter-chromosomal interactions. Previous studies have shown that inter-chromosomal interactions can bring multiple genes from different chromosomes into close physical proximity, thereby more easily enabling these genes to be co-regulated [32]. We therefore investigated the extent by which trans-eSNPs may regulate trans-eGenes located in different chromosomes via inter-chromosomal interactions. We observed that candidate trans-eSNP-eGene pairs display increased chromatin contact frequency compared with random inter-chromosomal contacts (Fig. 2F). Hence, in addition to cis-mediation, trans-eQTL associations can be partly driven by features of chromosomal conformation.
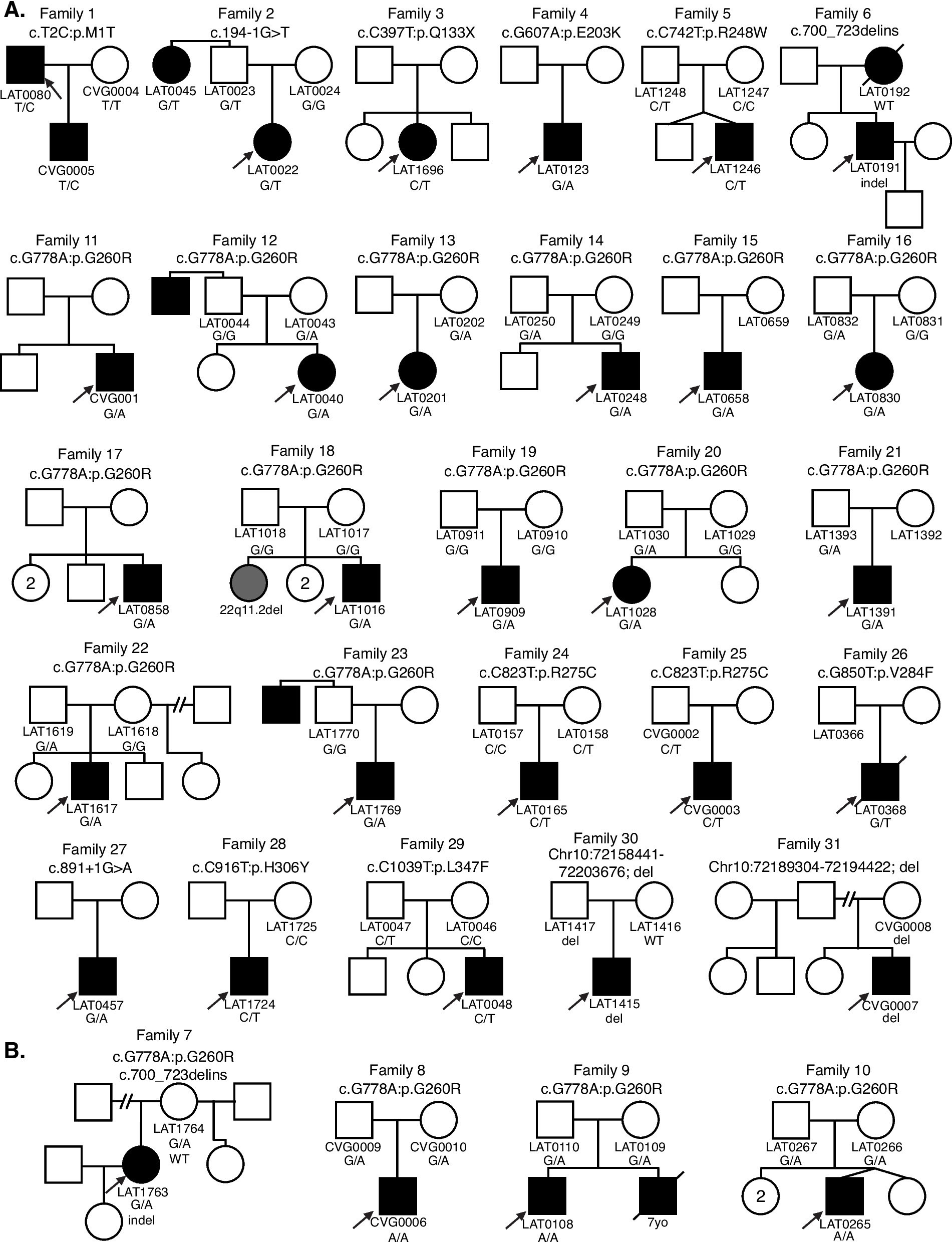
Trans-eQTL hotspotsTrans-regulators exert broad impacts on regulatory landscapes by affecting many downstream targets. Given the trans-regulatory properties of trans-eQTLs, some trans-eSNPs may affect multiple genes, thereby forming “trans-eQTL hotspots.” We defined such trans-eQTL hotspots as candidate trans-eSNPs that affect three or more genes (Fig. 3A). In total, we detected 382 trans-eQTL hotspots. Because trans-eGenes for a given trans-eQTL hotspot are regulated by the same SNP, we expect that they might generally be co-regulated. Indeed, we found that trans-eGenes grouped by hotspots are significantly more co-regulated compared to expression-level-matched random controls (Fig. 3B).
Fig. 3
Trans-eQTL hotspots. A Trans-eQTL hotspots represent trans-eSNPs that exhibit associations with at least three trans-eGenes. B Trans-eGenes associated with a shared trans-eSNP tend to be co-regulated. C Examples of trans-eQTL hotspots. The cis-eGenes are shown in orange boxes, and their respective trans-eGenes are shown in blue boxes. The red triangles are used to schematically designate the proximal relationship between the cis-eSNP and cis-eGene. D Cell-type-specific TF-target gene relationships detected by our mediator-candidate-trans-cis-QTL network
One of the identified trans-eQTL hotspots consists of three trans-eGenes (MAGEE2, CYBRD1, ZNF252), which are distributed across the genome and are regulated by a trans-eSNP (chr16:11394372; Fig. 3C). Notably, this trans-eSNP was a cis-eQTL for RMI2, a gene associated with genome instability and Bloom syndrome [33]. In another example, the cis-eSNP for RBM6 (chr3:50257020), an RNA-binding protein, was associated with three trans-eGenes (MAGEE2, MDH1B, AMACR, Fig. 3C). Collectively, these results suggest that multiple biological processes (such as genome instability and RNA processing) may exert broad impacts on gene regulation via trans-regulatory mechanisms.
Cell-type gene regulatory effects of candidate trans-eQTLs and mediatorsBecause candidate trans-eQTLs were defined from the brain homogenate and lack cell-type specificity, we evaluated cell-type-specific trans-regulatory effects from our mediators to trans-genes. For instance, our mediation analysis indicated that retinoic acid-related orphan receptor alpha (RORA, a nuclear receptor TF) is a mediator of the trans-eGene RNASEL, which encodes mammalian endoribonuclease. Based on single-cell multi-omics data, we found that RORA regulates the gene RNASEL specifically in neuronal cell types [29]. As shown in Fig. 3D, cellular expression levels of RORA and RNASEL show high Pearson correlation coefficients in several neuronal cell types, especially in inhibitory types, such as In6b (r = 0.831), In8 (r = 0.790), Ex9 (r = 0.746), and In6a (r = 0.742), compared to glial cell types such as microglia (r = 0.6436) and oligodendrocytes (r = 0.492).
Both of these genes have been implicated in disorders of the brain. For example, the overactivation of RNASEL may contribute to neurodevelopmental and inflammatory genetic disorders such as Aicardi–Goutières syndrome [34]. The expression of RNASEL may increase as a result of NMDA receptor activation in cortical neurons by glutamate, thus resulting in degradation of RNA molecules. Furthermore, degradation of mitochondrial RNA by RNASEL may contribute to neuronal death overall [35].
Reduced levels of the nuclear receptor TF RORA have been observed in the prefrontal cortex and cerebellar neurons of individuals with autism spectrum disorder (ASD) [36], and retinoic acid signaling pathways have been reported to be disrupted in ASD-afflicted individuals [37]. The decreased expression of RORA impacts the regulation of its target genes in ASD-afflicted individuals (several of which are ASD-relevant genes) and is associated with the pathobiology of ASD, such as with decreases in neuronal differentiation and survival, poorer synaptic transmission and neuroplasticity, diminished cognition and spatial learning, memory impairment, and disrupted development of the cortex and cerebellum [36]. Furthermore, studies have found that RORA is involved in the differentiation of Purkinje cells, development of the cerebellum region, protection of neurons against oxidative stress, circadian clock regulation, and suppression of inflammatory processes [36].
Candidate trans-eQTLs identify novel disease mechanismsIt has been proposed that trans-eQTLs explain 60–90% of gene expression heritability [38, 39]. However, functional annotation of GWAS variants largely relies on the use of cis-eQTLs, which may miss key biological underpinnings of human traits and diseases. Trans-eQTLs may provide novel insights into the biological mechanisms underlying psychiatric illnesses. Therefore, we performed colocalization analysis [19] between SCZ GWAS [40] and candidate trans-eQTLs to unveil previously uncharacterized SCZ-associated biological pathways driven by trans-regulatory mechanisms (“Methods”). We then compared these results with the colocalization results between SCZ GWAS and cis-eQTLs. We found that some loci only colocalized with cis- or candidate trans-eQTLs, although several colocalized with both.
In total, we found that candidate trans-eQTLs could explain 55 out of 142 SCZ-associated genome-wide significant (GWS) loci (Fig. 4A). In contrast, cis-eQTLs annotated 78 GWS loci (Fig. 4A). 32 GWS loci colocalized with both cis- and candidate trans-eQTLs, suggesting that a subset of SCZ loci may exert their effects via multiple regulatory mechanisms. Furthermore, 23 GWS loci colocalized only with candidate trans-eQTLs but not with cis-eQTLs, suggesting that trans-eQTLs may provide regulatory mechanisms for previously unexplained loci.
Fig. 4
Candidate trans-eQTLs identify novel SCZ risk genes and biological pathways. A The numbers of SCZ GWS loci annotated by cis- and candidate trans-eQTLs. SCZ cis-eGenes do not overlap with SCZ trans-eGenes. B Trans-eGenes that colocalize with SCZ risk genes exhibit greater network centrality. C Cellular expression profiles of SCZ-associated cis- and trans-eGenes. D A SCZ GWS locus colocalizes with candidate trans-eQTLs for CENPX (left). This GWS locus is a cis-eQTL for RPS17, a cis-mediator of CENPX (right)
A colocalization analysis resulted in 90 and 282 SCZ-associated trans- and cis-eGenes, respectively; we refer to these as SCZ-(trans/cis)-eGenes (Fig. 4A). As expected, none of the SCZ-cis- and SCZ-trans-eGenes overlapped, demonstrating that candidate trans-eQTLs can pinpoint distinct SCZ-associated genes and biological pathways. In particular, SCZ trans-eGenes were found to be enriched for JUN kinase activity (FDR=0.0056), a signaling pathway involved in neuronal apoptosis, neurite outgrowth, and dendrite arborization [41].
Notably, two of the 90 SCZ trans-eGenes (AKAP11, SETD1A) also harbor SCZ-associated rare variants (de novo loss-of-function [LoF] variation) [23] (Fisher’s exact test, P=9.6×10−3, odds ratio [OR]=14.62, 95% confidence interval [CI]=1.67–59.06; see Additional file 6: Fig. S7 for colocalization between SETD1A candidate trans-eQTLs and SCZ GWAS). This contrasts with the 282 SCZ cis-eGenes, none of which overlapped with the genes that harbor SCZ-associated rare de novo LoF variants. This result lends support to the omnigenic hypothesis [38], which posits that genes can be divided into core genes that directly affect the disease-related biological processes and peripheral genes that regulate these core genes. Core genes are likely to be affected by rare variants with large effect sizes, suggesting that these two SCZ trans-eGenes are likely core genes. In addition, the hypothesis suggests that core genes are likely to be targeted by trans-regulatory mechanisms, which is consistent with these two genes being trans-eGenes of SCZ rare variants that are mediated by peripheral genes.
To provide additional support for the omnigenic hypothesis, we explored the network properties of SCZ trans-eGenes. Core genes are thought to be enriched for network hubs [42]. Therefore, we measured module membership (kME; a measure of network centrality) of SCZ-eGenes in brain co-expression networks constructed from the same samples [24]. SCZ trans-eGenes showed significantly higher kME values than did cis-eGenes or brain-expressed genes in general (Fig. 4B, two-sample Wilcoxon test: cis vs. trans, P=9.64×10−7; all vs. trans, P=2.87×10−9; all vs. cis, P=9.9×10−2). These results demonstrate that SCZ trans-eGenes are characterized by greater network connectivity.
We found that a significant portion of SCZ-associated trans- and cis-eGenes were differentially regulated in brain tissue of SCZ-affected individuals [42] (Fisher’s exact test: trans, P=9.4×10−3, OR=1.87, 95% CI=1.14–2.98; cis, P=3.5×10−2, OR=1.38, 95% CI=1.01–1.85). However, they were enriched in different SCZ-associated co-expression networks. SCZ trans-eGenes showed selective enrichment in the SCZ-associated gene module 7 (geneM7, beta=0.0033, FDR=0.011), a neuronal module involved in synaptic vesicle formation that exhibits elevated expression signatures in SCZ (Fisher’s exact test: P=8.02×10−5, FDR=0.0014, OR=5.52, 95% CI=2.42–11.10). In contrast, SCZ cis-eGenes were only nominally enriched in gene module 8 (geneM8, beta=-0.0030, FDR=0.017), a neuronal module downregulated in SCZ (Fisher’s exact test: P=1.8×10−2, FDR=0.53, OR=2.31, 95% CI=1.13–4.24). Therefore, SCZ-associated trans- and cis-eGenes may account for different expression signatures of SCZ.
Cell-type expression profiles of SCZ-eGenes illustrated potential cell types that contribute to distinct expression features (Fig. 4C). Both SCZ-associated trans- and cis-eGenes were highly expressed in neurons, which is consistent with previous findings that neurons are the primary cell type underlying SCZ etiology [27, 43,44,45,46]. However, SCZ cis-eGenes exhibited relatively higher expression in lower-layer neurons (Ex7-8), while SCZ trans-eGenes showed upper-to-lower-layer gradient expression. Furthermore, while SCZ cis-eGenes were relatively depleted in inhibitory neurons, SCZ trans-eGenes were highly expressed in parvalbumin-expressing GABAergic interneurons (In6). One example of a SCZ-trans-eGene is CENPX, which is associated with kinetochore assembly and DNA damage repair [47]. SCZ GWAS colocalized with candidate trans-eQTLs for CENPX, which was mediated by cis-eQTLs for RPS17, a gene that encodes a ribosomal protein (Fig. 4D). Ribosomal proteins can affect translation efficiency and mRNA stability. This result suggests that an SCZ GWAS SNP may affect the nearby gene RPS17, which potentially alters the mRNA stability and in turn regulates CENPX, a gene located in a different chromosome. Together, these results demonstrate how the integration of GWAS variants, cis-eQTLs, and candidate trans-eQTLs can enhance our mechanistic understanding of disease etiology.
留言 (0)