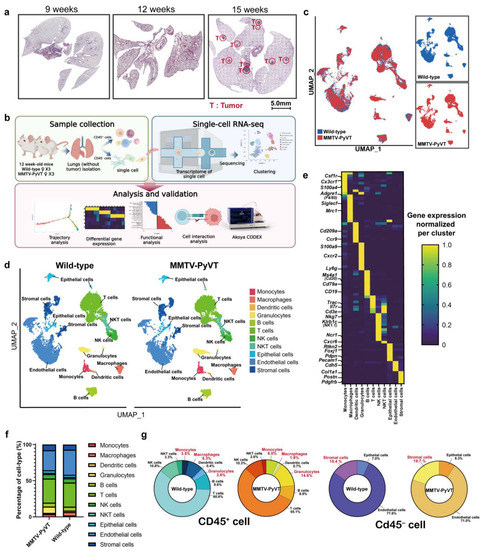
1. IntroductionThe incidence of bladder cancer (BC) ranks eleventh among all malignant tumors in the world [
1]. BC is divided into two types, the majority being non-muscle-invasive diseases (90%) and the minority being muscle-invasive diseases [
2,
3]. Non-muscle-invasive BC frequently recurs (47%) [
4], which greatly affects patients’ quality of life. Furthermore, muscle-invasive BC has a poor prognosis and a five-year survival rate of
5]. Despite advances in chemotherapy and surgery, the prognosis has not changed significantly, and new entry points for treatment options are urgently needed [
6]. Therefore, it is important for BC diagnosis and prognosis to discover novel biomarkers. SCL/TAL1 interrupting locus (STIL), a key regulator involved in the regulation of centriole duplication, is an important checkpoint protein in mitosis [
7]. Chromosomal instability caused by ectopic centriolar amplification is the main feature of human cancer [
8]. Elevated expression of STIL has been reported in various cancers, such as colorectal cancer, pancreatic cancer, gastric cancer, prostate cancer, lung cancer, and nasopharyngeal carcinoma [
9,
10,
11,
12,
13,
14]. STIL promotes the development of many cancers; however, there is no detailed investigation of STIL in BC.
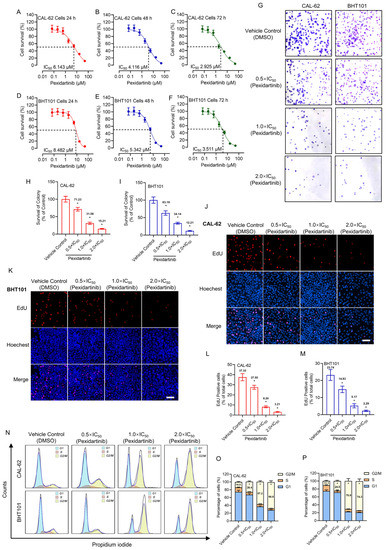
In our study, we found that STIL was abnormally expressed in BC patients and predicted a poor prognosis. Moreover, STIL knockout markedly blocked proliferation and migration of BC cells in vitro and inhibited proliferation of tumor in vivo, and triggered cell cycle arrest in the G0/G1 phase. Mechanistically, STIL promoted the PI3K/AKT/mTOR pathway and increased c-myc expression, thereby facilitating BC progression.
2. Materials and Methods 2.1. Tissue Microarray and Immunohistochemistry
TMA (tissue microarrays) (product code: HBlaU079Su01, Outdo Biotech, Shanghai, China) included 63 BC tissues and 16 paracancer tissues. The samples were collected from May 2007 to January 2011. TMA slices were dehydrated with different concentrations of gradient alcohol. After being made transparent with a transparent agent (xylene), the slices were soaked in 3 pots of paraffin at 60 °C one by one for 1 h and finally embedded and sectioned. Antigen repair was performed in an electrochromic oven with a repair solution (0.01 mM EDTA buffer, pH 9.0). Three-percent hydrogen peroxide was added to block endogenous peroxidase, and goat serum was blocked at room temperature for 30 min. Then, the slices were incubated overnight (12 h) with a primary antibody. Next, the slices were incubated with the second antibody at 37 °C for 30 min, and then with a freshly prepared DAB chromogenic solution. After redyeing with hematoxylin, the slices were dehydrated, sealed, scanned, and photographed under a microscope. Immunohistochemical scores were performed independently by two experienced pathologists. In general, the expression of STIL was evaluated on the criteria of the intensity and positive staining rate of immunostaining of the tumor tissue. The positive signal was brownish yellow or brown. The staining intensity was scored as 0, 1, 2, and 3. The positive staining rate score was defined as 0 (0%), 1 (1–25%), 2 (26–50%), 3 (50–75%), and 4 (75–100%). Histoscore was calculated by multiplying the staining intensity score and the staining positive rate score.
2.2. Cell Culture
The human BC cell lines include the UMUC3, EJ, J82, T24, and SCABAR cell lines, HEK293T cell line, and normal human bladder cell line SV-HUC-1. These cells were donated by the Stem Cell Bank, CASS, China. One hundred U/mL penicillin, 10% fetal bovine serum (Invitrogen, Shanghai, China), and 0.1 g/mL streptomycin sulfate were added to RPMI medium 1640 (Invitrogen, Shanghai, China) or DMEM high glucose medium (Gibco, Shanghai, China). SV-HUC-1, EJ, and T24 cells were cultured in mixed RPMI medium 1640; UMUC3, J82, SCABAR, and HEK293T cells were cultured in mixed DMEM high-glucose medium. All cells were incubated at 37 °C with 5% CO2 and 95% air.
2.3. STIL Knockout in UMUC3 and T24 Cells
We performed STIL knockout in UMUC3 and T24 cells with the CRISPR-Cas9 system. In this system, small guide RNAs (sgRNAs) of STIL were cloned into lenti-v2 (Addgene, 92062, Shanghai, China). HEK293T cells were co-transfected with recombinant lenti-CRISPR-v2 and package plasmid to generate lentivirus for 48 h. The supernatant was collected and added to UMUC3 and T24 cells, and incubated for 36 h; subsequently, 1000 ng/mL puromycin was added for 4 days. We used a limited dilution method to obtain the cloned cells (UMUC3 and T24 cells were immobilized on five 96-well plates and grown for 3 weeks). We screened monoclonal cells by Western blotting to obtain accurate STIL knockout cells. The sgRNA sequences were sgRNA-F, 5′-caccGGGGTTATTTCTAGGCATTC-3′; sgRNA-R: 5′-aaacGAATGCCTAGAAATAACCCC-3′.
2.4. CCK-8 Assay
The cells with STIL knockout were inoculated into 96-well plates with 2 × 103 cells per well. The absorbance of 96-well plates was measured on days 1, 2, 3, 4, 5, and 6, respectively. Ten μL of CCK-8 solution (BS350A, biosharp, Hefei, China) was added to each well and cultured in a cell incubator for 3 h. The optical density was measured at 450 nm using a multifunctional enzyme marker (PE Enspire, PerkinElmer, Singapore).
2.5. Cell Colony Formation
The cells were inoculated into 6-well plates at a density of 1.2 × 103 cells per well and cultured for about 3 weeks until clearly visible to the naked eye. Finally, they were dyed with 0.3% crystal violet (dissolved by anhydrous ethanol), washed gently with water, and photographed.
2.6. Transwell Migration Assays
Medium containing 20% fetal bovine serum was added to the lower chamber of the 24-well plates, and 5 × 104 cells with serum-free medium were added to the upper chamber of a transwell chamber (Thermo Fisher Scientific, Shanghai, China) to culture for 24 h. The migrated cells were fixed with 4% paraformaldehyde (biosharp, Shanghai, China), stained with 0.3% crystal violet, photographed, and counted with an inverted microscope.
2.7. Soft Agar Assay
One-point-four-percent low-melting-point agarose (Promega, Madison, WI, USA) was added to 6-well plates and solidified at room temperature. Ten thousand cells were mixed with 0.7% low-melting-point agarose and overlaid on 1.4% low-melting-point agarose. After solidifying at room temperature again, they were put into an incubator and incubated for about 4 weeks before being photographed under a microscope.
2.8. Cell Cycle Assay
We used a cell cycle staining kit (Multisciences, 70-CCS012, Shanghai, China). After collecting 5 × 105 cells, the cells were washed with PBS. Then, 1 mL of DNA staining solution and 10 μL of osmotic solution were added to the cells. After incubating at room temperature for 30 min, the cells were detected on a flow cytometer (CytoFlex S, Beckman Coulter, Wuhan, China) with the lowest flow rate.
2.9. The EDU (5-Ethynyl-2-deoxyuridine) Assay
EDU cell proliferation detection kit (C0071S, Shanghai, China) was purchased from Beyotime. Cells were incubated with EDU working solution for 2 h, then fixed with 4% paraformaldehyde and treated with permeabilization solution (P0097, Immunostaining Strong Permeabilization Solution, Beyotime, Shanghai, China) for 30 min at room temperature. According to the protocol of the manufacturer, cells were incubated in click reaction solution (CuSO4, Click Reaction Buffer, Click Additive Solution, Azide 488) at room temperature in the dark for 30 min. Nuclei staining was conducted with Hoechst (33342, Beyotime, Shanghai, China) reagent. After staining, the cells were observed under a confocal fluorescence microscope or prepared as cell suspension samples for flow cytometry.
2.10. Immunofluorescence Assay
The Ki67 cell proliferation kit (E607238-0100, sangon, Shanghai, China) was used to measure cell proliferation. For specific steps, please refer to the instruction manual. Briefly, cells were incubated with the diluted primary antibody Ki67 overnight at 4 °C and incubated with the fluorescent-labeled secondary antibody at 37 °C for 30 min. Nuclei were then counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Finally, immunofluorescence staining was observed under an inverted fluorescence microscope.
2.11. RT-qPCRRNA was extracted from cells using trizol reagent (Invitrogen, Shanghai, China); then, reverse transcription was performed using the cDNA reverse transcription kit (Toyobo Life Science, Shanghai, China) to synthesize cDNA. RT-qPCR was performed using the SYBR Green PCR kit (TSE201, Tsingke Biotechnology, Wuhan, China) and a Bio-Rad CFX96 PCR system to detect CT values. Primer sequences are in
Supplementary Table S1. 2.12. Western BlottingProtease inhibitors were added to the RIPA buffer (Beyotime, Wuhan, China) to lyse cells on ice and eventually extract proteins. The proteins were electrophoretic in 8–15% SDS-PAGE gel and transferred to PVDF membrane (IPVH00010, Sigma-Aldrich, Shanghai, China), after which they were sealed with blocking buffer (P30500, Ncm Biotech, Suzhou China) for 15 min. The membrane was incubated overnight in the designated primary antibody at 4 °C. After washing with TBST, the membrane was incubated with the corresponding secondary antibody at room temperature for 1 h. Subsequently, the membrane was imaged on a chemiluminescence image analysis system (Tanon 5200, Wuhan, China). STIL antibodies were purchased from Santa (SC-271910, Dallas, TX, USA). Original blots see
File S1. 2.13. Tumor Xenografts
The experimental plan was approved by the Animal Biosafety Level III Laboratory of Wuhan University. We injected cells via syringe into the backs of nude mice living in a pathogen-free environment (6 × 106 cells/nude). The nude mice were purchased from China Shanghai Laboratory Animal Research Center. Tumor volume was measured every 3 days after surgery and calculated by the formula: length × width × height × π/6. On the 26th postoperative day, the tumors of mice were collected, photographed, and weighed.
2.14. Statistical Analysis
SPSS 25.0 was used for statistical analysis. All experiments were performed at least 3 times. One-way ANOVA was used for three or more groups. A two-sided Student’s t-test was used for the significance of the difference between the two groups. p < 0.05 was considered statistically significant (* p < 0.05, ** p < 0.001, and *** p < 0.001).
2.15. RNA-Seq Analysis of N-butyl-N-(4-hydroxybutyl) Nitrosamine (BBN)-Treated Bladder Cancer Mouse ModelsWe obtained the raw sequencing data from the NCBI SRA Database with Bioproject: PRJNA587619 [
15]. STAR was used to align RNA sequencing reads [
16], and featureCounts was used for quantification of gene and transcript levels [
17]. And differentially expressed genes were screened between the BBN-treated group and control group by R package Deseq2 [
18]. 4. DiscussionPrevious studies have shown that STIL causes abnormal centriole expansion, which, in turn, leads to chromosomal instability [
7]. Chromosomal instability is the hallmark of many cancers [
8]. STIL is considered to be an oncogene whose expression is elevated in many types of cancers [
9,
10,
11,
12,
13]. However, there is little in the literature on the question of STIL in BC. Our study found that the mRNA expression levels of STIL were significantly elevated and were positively correlated with the cycle-related gene (CCNB1, CDK1, CCNA2, CCNB2, and CCNE2) in BC. In addition, BC patients with high expression of STIL had poor prognoses. Demonstrated by microarray detection and immunohistochemistry, we confirmed an increase in STIL expression in clinical specimens. Previous studies have suggested that STIL is critical for cancer cell migration [
19]. Our experimental results show that STIL has similar results in BC: STIL knockout inhibited BC cells proliferation and migration in vitro and blocked proliferation of tumor in vivo. These results strongly showed that STIL is related to the development and occurrence of BC. STIL is a cell-proliferation-related gene involved in cell cycle regulation [
20]. We analyzed cell proliferation by detecting cell proliferation markers (EDU, Ki67), as well as observing changes in cell cycle and cycle-related proteins. According to previous studies, the ability of the G0/G1 phase to enter the S phase is mainly determined by CDKs, including CDK2/4/6 [
21,
22]. Cyclin D and CDK4/6 are highly related and readily form conjugates. By activating CDK2, the cyclin D-CDK4/6 complex promotes DNA replication and cell proliferation [
22]. In many tumors, reductions in CDK2/4/6 and cyclin D1 often coexist with G0/G1 cell cycle arrest [
23,
24,
25]. And some studies have shown that STIL is closely related to G1 phase arrest [
26]. A similar phenomenon emerged in our study: STIL knockout resulted in cell cycle arrest in the G0/G1 phase in BC cells. At the protein level, CDK2/4/6 and cyclin D1 were decreased in STIL knockout BC cells. STIL knockout showed different blockade phases in different tumor types, and reduced the proliferation of cervical and colon cancer cells by inhibiting Cyclin B1/CDK1, which, in turn, induced cell cycle arrest in the G2/M phase [
11,
27,
28]. To explore the effects of STIL on proliferation in vivo, we used a xenograft model and confirmed that STIL knockout inhibited the growth of xenograft tumor cells. Our RNA sequencing results and gene set enrichment analysis (GSEA) showed a significant decrease in PI3K/AKT/MTOR pathway enrichment and c-myc target enrichment after STIL knockout. Heat map analysis showed that the downstream molecules of c-myc were significantly down-regulated in STIL knockout BC cells. RT-qPCR and Western blotting were used to confirm that the downstream p-PI3K/p-AKT/p-mTOR/c-myc pathway was down-regulated after STIL knockout. These results suggest that STIL plays a cycle-related gene vital role in BC through the PI3K/AKT/mTOR pathway and c-myc. Significant research has shown that the PI3K/AKT signaling pathway plays a significant role in promoting invasion, migration, proliferation, and other malignant characteristics of human cancers [
29,
30]. C-myc, as a proto-oncogene, is fundamental to regulating cell proliferation [
31,
32]. Several studies have documented that the activation of c-myc is mediated by the AKT/mTOR pathway [
33,
34,
35]. Furthermore, c-myc is up-regulated in many tumors and is important for proliferation [
36,
37]. Additionally, c-myc is crucial to the cell cycle progression of tumor cells [
38]. C-myc acts as a transcription factor to stimulate cell cycle progression and cell proliferation [
39,
40]. It has been proposed that c-myc regulates cell cycle progression through CDK2/4 and cyclin D1 [
41,
42]. These previous studies are consistent with our findings. Thus, we conclude that STIL reduces c-myc through the PI3K/AKT/ mTOR signaling pathway, leading to a decrease in CDK2/4/6 and cyclin D1, thereby causing cell cycle arrest in the G0/G1 phase and ultimately inhibiting cell proliferation. Therefore, in the future, it may be possible to inhibit tumor proliferation by inhibiting STIL, reducing c-myc, and arresting the G0/G1 phase. Additionally, PI3K/AKT/mTOR signal transduction is modulated by SC79 in BC cells, which significantly reduces the down-regulation of STIL knockout. We proved that changes in STIL drive fluctuations in c-myc, which affect the cell cycle. However, we still do not know the direct target of STIL; we will address this question in future research.
In summary, we are the first to confirm that STIL may promote development in BC. Our results suggest that STIL is strongly associated with prognosis in BC patients and mediates c-myc through the PI3K/AKT/ mTOR signaling pathway, ultimately promoting the BC cell cycle, proliferation, invasion, and metastasis. These findings may shed new light on the role of STIL in human malignancies and provide new targets for treating BC.

留言 (0)