
記住我
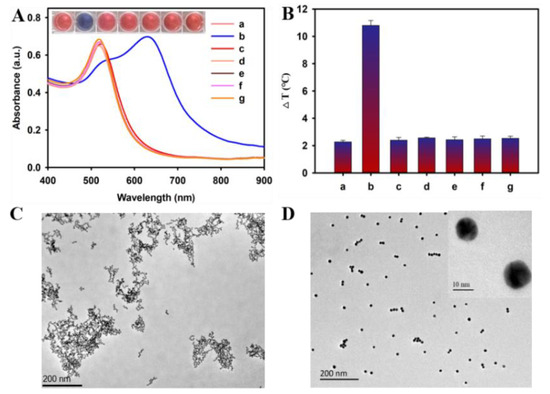

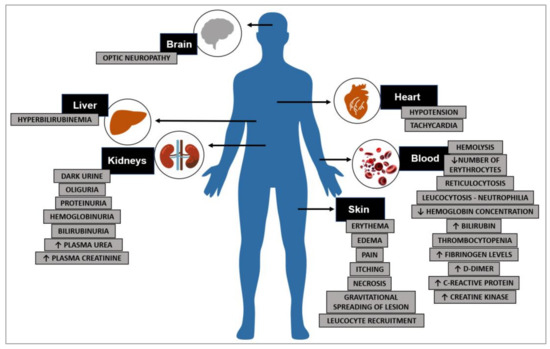
The in vivo manifestations of ricin toxicity are highly dependent upon the route of administration because of ricin’s promiscuous cell-binding abilities. Nevertheless, there are important commonalities to the response, notably apoptosis and inflammation. That parenteral administration of ricin would lead to a burst of pro-inflammatory cytokines within hours was predictable. That this would lead to fatal hypoglycemia was not.
To further understand the consequences of these changes, we have performed metabolite profiling analyses of the livers of ricin treated-mice and as a control, food-deprived animals. The results show that each treatment induces a metabolically distinct hypoglycemic state [48]. Understanding the metabolic causes, consequences, and response to hypoglycemia has direct relevance for the management of ricin intoxications, diabetes mellitus, and other metabolic syndromes. In the future we propose to extend these analyses to include insulin-induced hypoglycemia, because of the important adverse consequences of hypoglycemia on the long-term management of diabetes mellitus [62,63,64]. 4. Materials and Methods 4.1. MiceOutbred Swiss (Cr:NIH(S), Charles River Laboratories, Wilmington, MA, USA) mice were studied, including 164 females and 25 males. Outbred mice were chosen to avoid strain-specific genetic effects. The female to male imbalance resulted from different housing requirements. Outbred mice were between 2 and 5 months of age and weighed between 17–40 g. The mice were treated as follows: fasted (12 mice), injected with saline (34 mice) or ricin 30 µg/kg (124 mice), 15 µg/kg (1 mouse), 7.5 µg/kg (6 mice), 2.5µg/kg (6 mice), ricin A-chain 60 µg/kg (6 mice), or ricin B-chain 60 µg/kg (6 mice). Injections were ip (177 mice) or iv (12 mice). NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG, Jackson Labs, Bar Harbor, ME, USA) are defective in both adaptive and innate immunity, as well as in cytokine signaling. A total of 14 NSG mice were used: 8 females that were 10 months old, 3 females and 3 males that were 3–4 months old. The mice were 18–30 g and were injected with ricin 30 µg/kg. Fifty-six inbred BALB/c mice (colony breeders originated from Jackson Laboratory, and the colony was maintained at Montana State University’s Animal Resource Center), half male and half female, were studied. Inbred mice limit variability and the strain specific inflammatory response is well documented for BALB/c mice [65,66]. All of the mice were 4–11 weeks old, weighing 20–30 g when studied. BALB/c mice were injected with saline (16 mice) or ricin 30 µg/kg (40 mice). All strains of mice were monitored and euthanized at specific time points.Mice were housed in Tecniplast caging West Chester, PA, USA and individually ventilated with HEPA filters and aspen wood chips (Sanichips, P.J. Murphy’s, Montville, NJ, USA). For enrichment, the mice were given a red mouse house (Bioserve, Flemington, NJ, USA), a nestlet (Ancare, Bellmore, NY, USA, and EnviroDry (Shephard Specialty Papers, Amherst, MA, USA). All cage components were autoclaved prior to use. The mice were fed with irradiated PicoLab Rodent Diet 20 (PMI Nutritional, North Arden Hills, MN, USA). The animals had access to water and feed for the entire experiment, unless otherwise noted. The ambient air temperature was 70–72 °F with a minimum of 30% humidity. The light was on from 5 a.m. to 7 p.m. and off for the remainder of the day for a 14/10 cycle. The female mice were housed 4–5 mice per cage and the males 1–4, depending upon age and aggressiveness.Previous experiments demonstrated the degree of experimental variability [21]. Based on these results, power analysis was performed to determine the group size. It was determined that 6–8 animals per group provides a >90% probability of obtaining statistically significant differences (pAnimals were bled > 1 d prior to ricin injection into a heparinized tube or a blood glucose test strip. The mice were bled from the saphenous, facial, or submandibular vein, at the discretion of the animal technician. The mice were injected ip, unless otherwise noted, with 10 µL of volume per gram body weight, via a 27 ga 3/8″ needle. The animals were monitored by AALAS-certified laboratory animal technicians every 20–30 min for the initial 2 h post injection, and every 2 h to 14 h thereafter. Any mice demonstrating signs of distress, pain or suffering were euthanized. The specific criteria for early euthanasia were loss of appetite, markedly diminished activity levels, lethargy, neurologic signs, unexpected behaviors, mottled fur, hunching, difficulty breathing, respiratory sounds, or blood glucose < 25 mg/dL.
At the time of sacrifice, a terminal bleed was performed by intracardiac puncture while under anesthesia. Animals were anesthetized using isoflurane by inhalation, dosing was accomplished by adjusting the position of the nose cone, and anesthesia was confirmed by loss of toe withdrawal reflex. Isoflurane was administered for the duration of the intracardiac bleed and continued until euthanasia. Euthanasia was performed by a cervical dislocation and thoracic incision. The liver, skeletal muscle, pancreas, and plasma were then removed from the mice.
4.2. Abs, Reagents, and Test KitsRicin holotoxin (5 mg/mL, Ricin Agglutinin II (RCA 60)*, L-1090), ricin A chain (L-1190), and ricin B chain (L-1290) were obtained from Vector Laboratories. The ricin holotoxin (5 mg/mL) was in a saline solution containing 10 mM sodium phosphate, 0.15 M NaCl, pH 7.8, 0.08% sodium azide, 5 mM galactose, then diluted further in PBS prior to use. STZ (Sigma, St. Louis, MO USA) was dissolved immediately prior to use in citrate buffer at pH 4.5 to 0.1 M or 0.02 M. Fluorescent Abs to murine cell surface markers were obtained from eBiosciences (San Diego, CA, USA): PE-Cy7-anti-CD45R/B220, PE-hamster anti-CD3e, and APC-rat anti-CD14 (clone rmC5-3). Similarly labeled isotype matched controls were also obtained from eBiosciences. Abs to detect phosphorylated and unphosphorylated members of the insulin signaling pathway were purchased from Santa Cruz Biotechnology: phospho-IRS-1, IRS-1, phospho-AKT, AKT, phospho-GSK-3β and GSK-3 β. Anti-ricin mAbs used in antigen-capture ELISA were produced from hybridoma cell lines, RAC18 (anti-A chain) and RBC11 (anti-B chain), created in our laboratory [18]. Cells were grown in RPMI1640 (Gibco, Billings, MT, USA) and 5% Ig-depleted FCS (HyClone, Logan, UT, USA) and purified on Protein A agarose (Sigma). RAC18 was conjugated to biotin with an EZ-Link Sulfo-NHS-LC-kiut (Thermo-Fisher). Streptavidin conjugated to horse radish peroxidase was obtained from GE Healthcare (Chicago, IL, USA). Abs for the detection of insulin and glucagon by immunostaining were rabbit Abs (Cell Signaling Technology, Beverly, MA, USA, mAb anti-insulin, polyclonal Ab anti-glucagon) and were detected with alkaline phosphatase-conjugated goat anti-rabbit IgG (Thermo-Fisher, Waltham, MA, USA). Binding was visualized using aminoethyl carbazole (Sigma). The kits used to detect specific biochemistry include: the glucagon-HS ELISA kit (Cosmo Bio USA, Carlsbad, CA, USA), L-lactate assay kit I (Eton Bioscience, San Diego, CA, USA), a colorimetric enzyme assay based on the reduction of tetrazolium salt in a NADH-coupled enzyme reaction to formazan, ketone body assay kit (Sigma), which measures 3-hydroxybutyric acid and acetoacetic acid using an enzymatic assay based on 3-hydroxybutyrate dehydrogenase catalyzed reactions, mouse ultrasensitive insulin ELISA kit (ALPCO, Salem, NH, USA), mouse G6Pc (Glucose-6-Phosphatase, Catalytic) ELISA kit (G-Bioscience, St. Louis, MO, USA), glucose-6-phosphate assay kit (Sigma Aldrich, St. Louis, MO, USA), and glycogen assay kit (Sigma Aldrich). The kits were employed following the manufacturers’ instructions and values measured relative to the standards enclosed with the kits. 4.3. Blood GlucoseBlood glucose was measured with a handheld glucometer (Accuchek Advantage, Roche Diagnostics Corp. (Indianapolis, IN, USA), and AgaMatrix. Inc. (Salem, NH, USA)). Blood (25 to 50 μL) was loaded onto the appropriate test strip for each glucometer and read 15 s later. Blood glucose was always measured prior to the experimental procedure and immediately prior to sacrifice. In addition, repeated measurements were made on 68 mice, between 1 and 48 h post ricin injection.
4.4. Measurement of Ricin in TissuesAn antigen capture ELISA was used to measure ricin holotoxin in the tissues [18,39]. Tissue samples were obtained at necropsy and immediately frozen. The samples were weighed and mixed with a 5× volume of ice-cold PBS 0.1% Triton X-100 (Sigma). The samples were homogenized for 60 s with a handheld tissue homogenizer, while on ice. Following sedimentation, the supernatants were tested for ricin. The wells of a 96-well round-bottom Immulon 2HB plate (Thermo Scientific) were coated with the mAb RBC-11, then blocked with milk buffer (PBS, non-fat dry milk, Tween 20, 10% Thimersol) for >1 d. The plates were rinsed and incubated with supernatants of tissue homogenates or ricin standards overnight, washed, and incubated with biotinylated mAb RAC 18. Following an overnight incubation, the plates were washed and avidin-conjugated HRP was added. Forty-five min later, the plates were washed, and binding was detected by incubation with tetramethylbenzidine (Sigma) for 15 min, then stopping the process with 1 M sulfuric acid, and reading at 450 nm on an automated plate reader (BioTek, Winooski, VT, USA). 4.5. Flow CytometrySingle cell suspensions of splenocytes were prepared and stained immediately post necropsy. The spleens were minced, passed through a nylon mesh (Falcon), and RBCs were removed with RBC lysis buffer (Sigma). The cells were resuspended in PBS/1%BSA/Azide. The cells (5 × 105) were stained in a total volume of 100 µL with an equal mixture of the following Abs: PE-Cy7-anti-CD45R/B220, PE-anti-CD3e, and APC anti-CD14. The cells were stained for 1 h on ice, washed with PBS, and fixed in PBS/2% paraformaldehyde. Analyses were performed on 10,000 cells gated by FSC and SSC using LSRII (BD Biosciences, San Jose, CA, USA). The data were analyzed with FlowJo software. A compensation algorithm was set using single stained cells and fluorescence-matched isotype controls (eBiosciences).
4.6. Multiplex AnalysesThe plasma obtained from the mice at the time of necropsy was tested for a panel of cytokines, mediators, and hormones using Milliplex Murine Map kits (MilliporeSigma, Burlington, MA, USA). Following the manufacturer’s instructions, recommended dilutions of plasma were mixed with multiplex beads. Beads and plasma were vortexed and centrifuged for 3 min. The following panels of multiplexed beads were used: bone1A (MBN1A-41K), cytokine/chemokine I (MPXMCYTO70KPMX), soluble cytokine receptor (MSCR-42K-PMX), IGF binding protein (MIGFBP-43K), cardiovascular disease I (MCVD1-77AK), cytokine/chemokine II (MPXMCYP2-PMX12), cytokine/chemokine panel III (MPXMCYP3-PMX6), CRP Single Plex (MCVD77K1CRP), IGF-1 single plex (RMIGF187K), endocrine (MENDO-75K), gut hormone (MGT-78K), and TGFß 1,2,3 (TGFB-64K-03). Each sample was run in duplicate. The samples were then assayed using a Bioplex 200 system (BioRad, Hercules, CA, USA). The concentrations of ligand were determined with the standards included in the bead sets.
4.7. Glucose Tolerance Test (GTT)We used a GTT to determine the ability of ricin-treated mice to clear glucose from the blood. A control group contained 6 mice and was injected with saline 24 h prior to GTT. Experimental groups of 4 mice each received ricin (30 µg/kg) 4 h, 6 h or 24 h prior to GTT. All animals were fasted 6 h prior to the beginning of the GTT, and water was allowed. Blood glucose was measured immediately prior to the start of the GTT. The mice were injected ip with 2 g glucose/kg body weight. Blood sugar was measured, using a glucometer as described previously, 15 m, 30 m, 60 m, and 120 m following glucose injection. Because blood sugar dropped with time post ricin injection, the area under the curve was calculated using a baseline normalized to blood sugar immediately prior to ricin injection.
4.8. ImmunoblottingSignaling by insulin was examined in the liver by immunoblotting as described elsewhere [55]. The liver was homogenized in cold lysis buffer (1% Nonidet P-40, 50 mM Hepes, pH 7.6, 250 mM NaCl, 10% glycerol, 1 mM EDTA, 20 mM β-glycerophosphate, 1 mM sodium orthovanadate, 1 mM sodium metabisulfite, 1 mM benzamidine hydrochloride, 10 μg/mL leupeptin, 20 μg/mL aprotinin, 1 mM phenylmethylsulfonyl fluoride). The supernatant was used for immunoblotting after centrifugation at 10,000× g for 10 min at 4 °C. Total protein (100 μg) in 50 μL of reducing sample buffer was used per lane. The product was resolved in 7% SDS-PAGE and transferred onto a polyvinylidene difluoride membrane for immunoblotting. Blotting of the membrane was conducted in a milk buffer with the first Ab for 1–24 h and HRP-conjugated secondary Ab for 30 m after. We used matched pairs of Abs recognizing the phosphorylated and total amounts of each protein: IRS-1, AKT, and GSK-3β. The phosphorylated signal and total signal strength were quantified with NIH Image J, and the ratio was calculated. The signals of treated samples are expressed as fold change over the untreated sample. 4.9. STZ-Induced DiabetesSTZ was used to induce diabetes as described elsewhere [44]. Prior to each STZ treatment, food was removed at 6 a.m. Four hours later, a fasting blood sugar was obtained, and STZ (40 mg/kg) was injected ip and the food was replaced. The mice received 5 daily doses of STZ. Fasting blood glucose was measured daily. When the blood glucose level was >250 mg/dL for 3 consecutive days, the mice were treated with ricin or saline. In the first experiment, three STZ-treated mice received ricin (30 or 15 µg/kg). The second experiment used 12 mice in 4 groups of 3. Two groups received STZ, two did not. The mice were injected with ricin (7.5 or 2.5 µg/kg). Blood glucose was measured twice daily post ricin. The mice were euthanized according to predetermined clinical criteria as outlined in the IACUC protocol and described above. 4.10. Microscopic AnalysesThe liver and pancreas were fixed in formalin, embedded in paraffin, sectioned and deparaffinized. For pathologic examination, the tissues were stained with H&E. IHC was performed with rabbit anti-insulin or anti-glucagon primary Abs, alkaline phosphatase-conjugated goat anti-rabbit IgG (Invitrogen, Waltham, MA, USA), and aminoethyl carbazole substrate (Sigma). IHC slides were counterstained with hematoxylin. Images were visualized using the built-in widefield capabilities of a Zeiss LSM 510 microscope and analyzed with Axiovision software (Zeiss, White Plains, NY, USA). Islets were identified by morphologic criteria using a 40× objective, a region of interest (ROI) drawn around the circumference of the islet, and measurement of the ROI’s area was calculated by the software.
4.11. Isolation of Islets from Intact PancreasPancreatic islets were isolated as described [67,68]. The mice were anesthetized with ketamine/xylazine. Collagenase (type 4, Worthington, 2 mg/mL) in Medium 199 (Gibco) was injected into the pancreatic duct in situ. The pancreas was immediately removed and placed in a glass screw cap vial on ice. The mouse was euthanized. Collagenase solution was added to each of the pancreases, which were placed in a shaking water bath at 37 °C for 15 min. RPMI 1640 medium with 10% FCS was added to stop the digestion, and the vial was placed on ice. The cells were washed centrifugally ×3 in RPMI 1640 + 10% FCS and resuspended in Ficoll-Paque (GE Healthcare) buffer. The cells were shaken to achieve suspension of pellets. RPMI1640 was layered on top of the Ficoll-Paque and centrifuged at 800× g for 20 min. The RPMI layer, interface, and the top of Ficoll-Paque layer were removed, washed, and placed in RPMI1640 + 10% FCS in petri dishes. With gentle swirling under a dissecting microscope, the islets were identified and collected. Isolated islets were saved in RNAlater (Qiagen, Hilden, Germany) or teased into single cell suspensions for immediate study. 4.12. Reverse Transcriptase, Quantitative PCR (RT-PCR)Relative changes in gene expression were quantified using real-time RT-PCR. The primers were designed to have similar annealing temperatures and are shown in Table 1. Total RNA was isolated from the liver or isolated pancreatic islets with an RNeasy mini kit (Qiagen) following DNase treatment. Quantitative real-time RT-PCRs were preformed using an iScript one-step RT-PCR kit with SYBR green (Bio-Rad, Hercules, CA, USA) on the iCycler MyiQ single-color real-time PCR detection system (Bio-Rad). A total of 10 ng of RNA was used per reaction and the transcript levels were normalized to GAPDH levels. The relative mRNA expression levels were calculated according to the comparative CT (∆∆CT) method as described by the manufacturer (User Bulletin #2, Applied Biosystems, Waltham, MA, USA). All of the reactions were performed in triplicate. 4.13. StatisticsAll of the statistics and graphs were generated using GraphPad Prism version 8.4.1 for macOS (GraphPad Software). The Shapiro–Wilk test and Brown–Forsythe test were performed on mice included in Figure 1 to validate the use of parametric tests throughout the manuscript. We have previously shown that blood glucose was the only parameter that significantly changed following ricin administration [21]. We reasoned that if the blood glucose data following ricin administration was parametric, the remaining data would be as well. Student’s t tests were performed when two groups were compared. When more than two groups were examined, one-way ANOVA with Dunnett’s multiple comparisons test was administered. The significance level was set at 0.05 for all statistical experiments and all of the groups were compared to the control mice. The mean ± SEM are shown in all graphical representations. If no error bars are visible, they are smaller than the symbol indicating the mean. Representative images are shown for tracing the islets of Langerhans, immunoblotting, and tissue staining experiments.
留言 (0)