
記住我
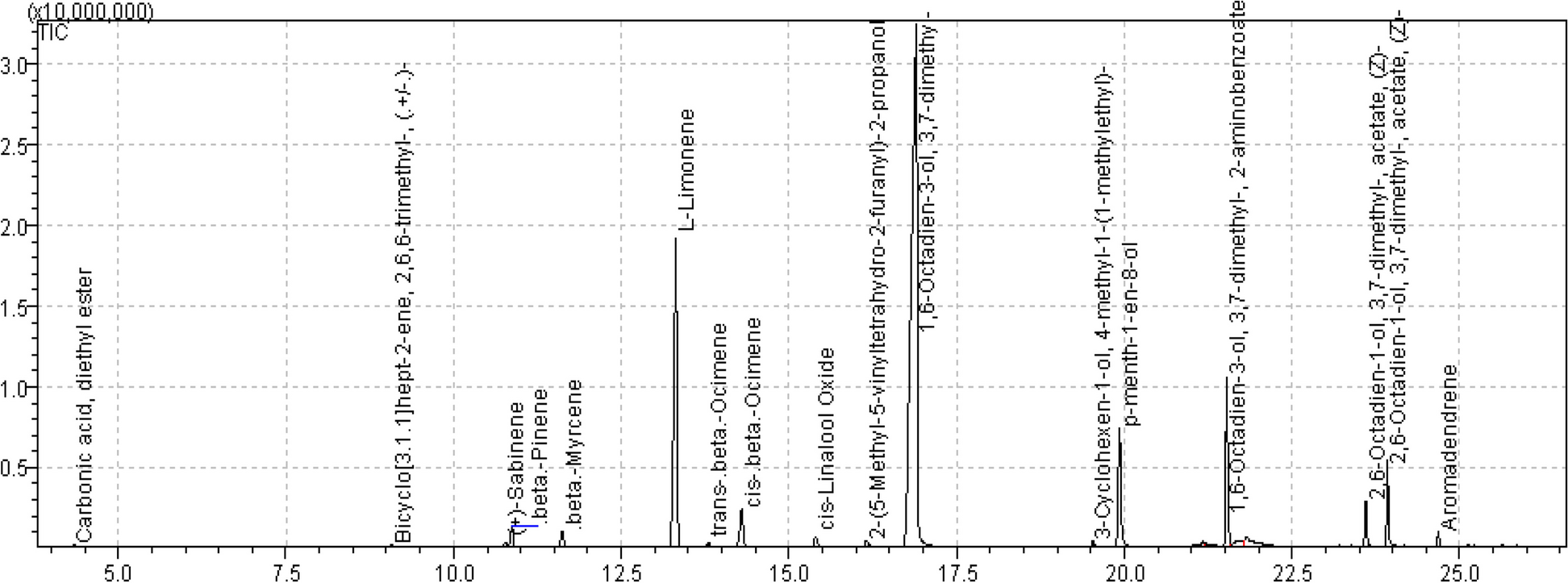
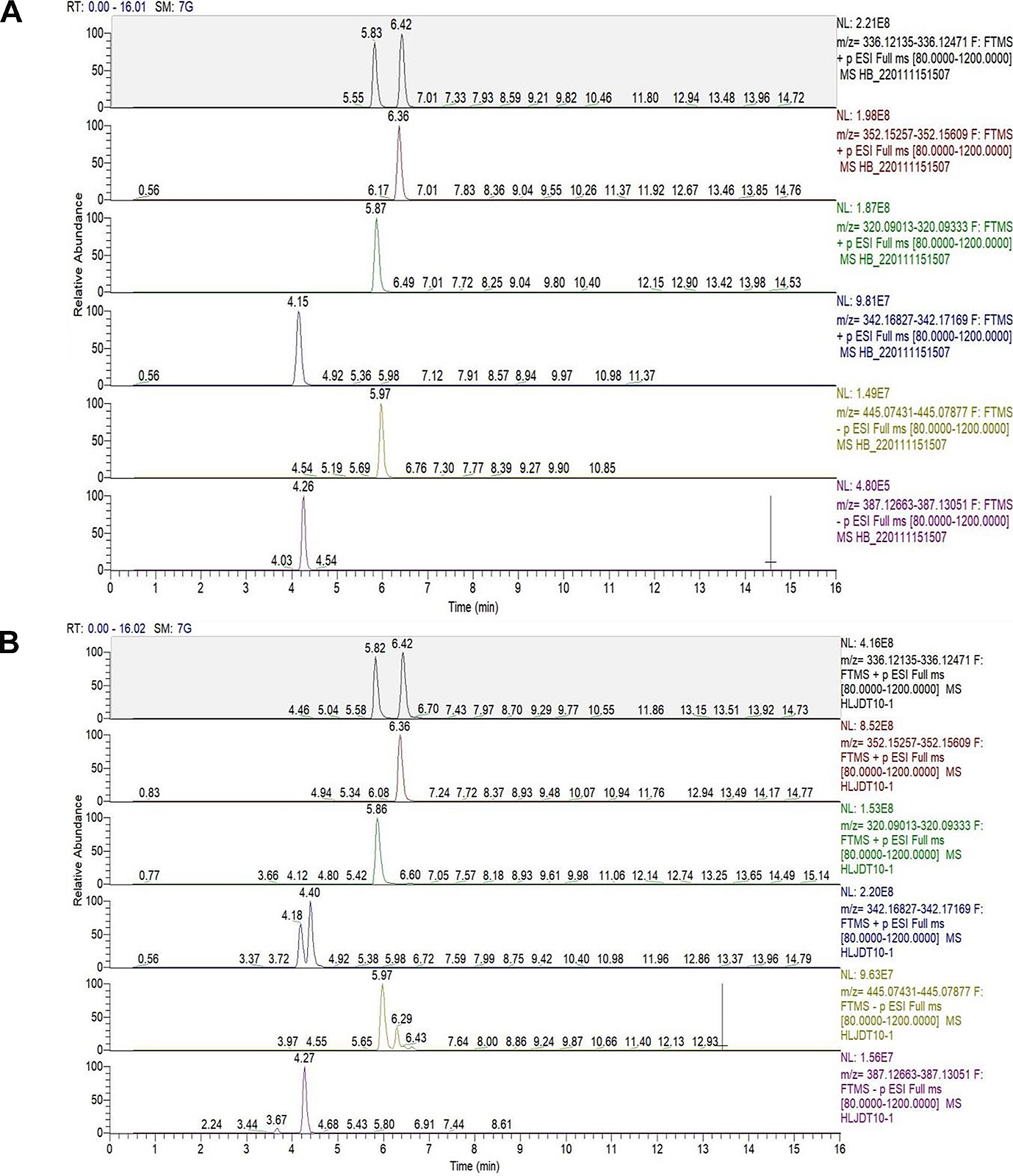
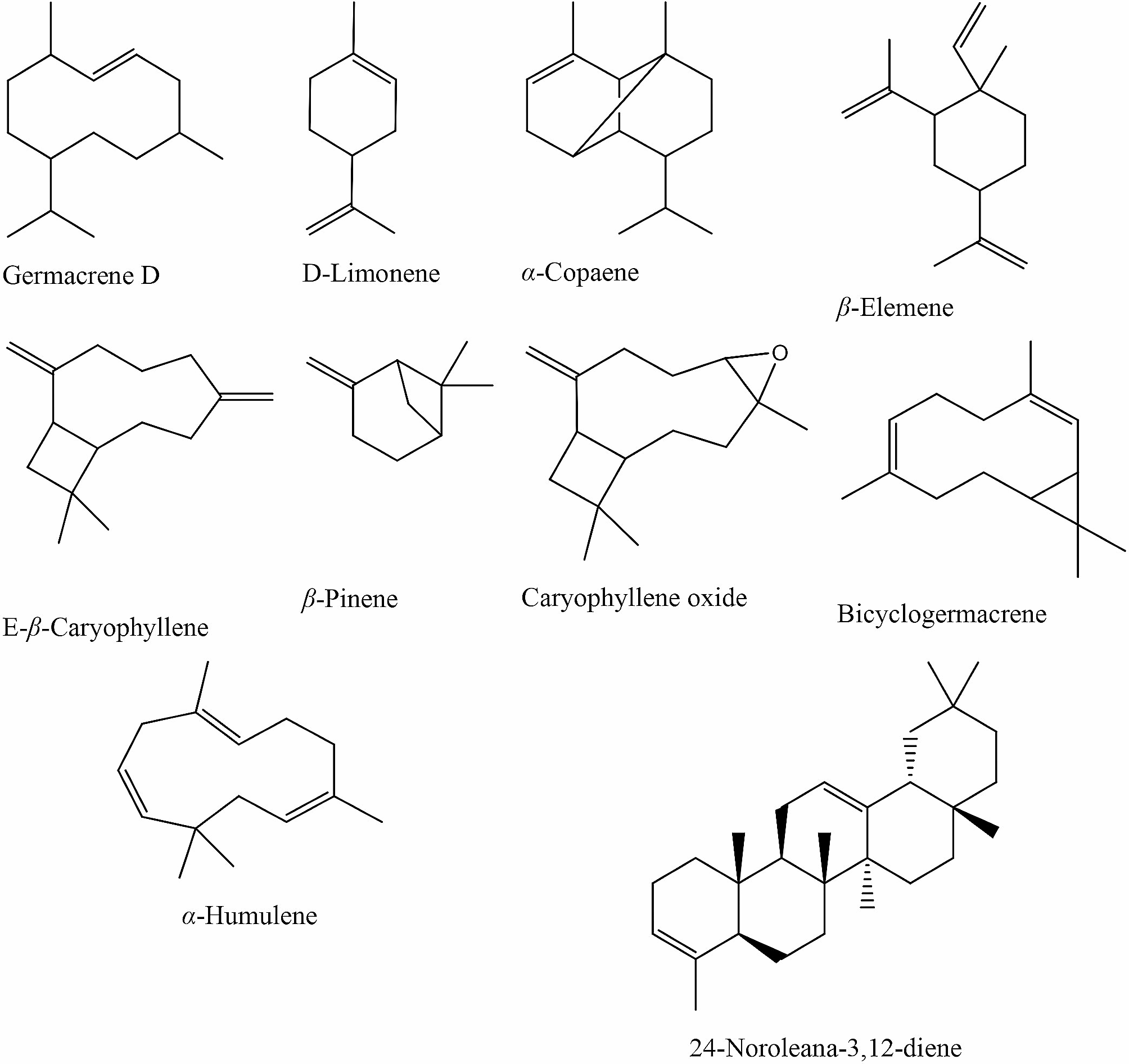

Seeds of C. anthelminticum were purchased from Hamdard Dawakhana, Saddar, Karachi. Identification was confirmed by the experts from the Department of Botany, University of Karachi, Karachi-75,270, Pakistan (voucher specimen: KU/BCH/SAQ/02).
ReagentsChloroform (Cat. no: 102447), glucose (Cat. no: D9434, dextrose), and trichloroacetic acid (TCA, Cat. no: T6399) were obtained from Merck & Co (New Jersey, US). Hexane (Cat. no: 296090, ethanol (Cat. no: V001229), DMSO (Cat. no: 276855), STZ (Cat. no: S0130), ketamine (Cat. no: K1884), xylazine (Cat. no: X1126), gliclazide (Cat. no: G2167), hydrochloric acid (HCl, Cat. no: 320331), hydrogen peroxide (H2O2, Cat. no: H1009), Ellman’s reagent (DTNB, 5,5′-dithiobis nitrobenzoic acid, Cat. no: D8130), glutathione (Cat. no: G4251), sodium azide (Cat. no: 13412), sodium arsenate (Cat. no: A6756), perchloric acid (Cat. no: V001526), hydroxylamine reagent (Cat. no: 159417), dichromate-acetic acid reagent (Cat. no: 223964), epinephrine (Cat. no: E4250), thiobarbituric acid (TBA, Cat. no: T5500) and TRIzol® Reagent (Cat.no: T9424) were procured from Sigma Aldrich Corp (St. Louis, MO, USA).
Extraction and fractionationThe purchased and identified seeds were thoroughly cleaned and weighed. The fixed oil of seeds was extracted using the Cold-press method at low temperature (below 50 °C) [35]. The extracted fixed oil was defatted twice with hexane to obtain hexane soluble fraction and the hexane insoluble residues were fractionated with chloroform to obtain chloroform soluble and insoluble fractions, the insoluble fraction was further fractionated with ethanol. The obtained fractions (hexane, chloroform, and ethanol) were subject to evaporation, and concentration using Büchi Rotavapor R-200 (62-65 °C). The fractions were kept in separate small vials labeled as HF (Hexane fraction), CF (Chloroform faction), and EF (Ethanol fraction) for further use (Fig. 1C).
Fig. 1
Schematic presentation of A experimental design of the study, B animal groups, and C cold press extraction of C. anthelminticum seed fixed oil (FO) followed by preparation of hexane (HF), chloroform (CF), and ethanol (CF) fractions of FO
Acute toxicity studyAn acute toxicity study of C. anthelminticum’s FO and its fractions were performed on Wistar Albino rats of both sexes, aged 6-10 weeks after they were fasted for 14-16 hours. The study was conducted in compliance with the Organization of Economic Co-Operation and Development (OECD) guideline 420 for testing of chemicals [36]. The FO of C. anthelminticum and its fractions were dissolved in 0.05% dimethyl sulfoxide (DMSO) and orally administered once at a dose of 500, 1000, 1500, and 2000 mg/kg, to their respective to groups rats (n = 6; 3 males, 3 females); whereas the control group only received 0.05% DMSO (1 mL/kg) as a vehicle. The animals were allowed free access to water and food, they were followed for 24 hours with strict observation in the initial 6 hours and daily thereafter for 2 weeks for signs of acute toxicity. Once daily for 14 days, the animals were observed for changes in physical appearance, behavior, mortality, (i.e. salivation, lethargy, etc.), and acute illness/injury. On the 15th day, animals were euthanized through intraperitoneal injections of Ketamine 60 mg/kg and Xylazine 7 mg/kg body weight [37]. The cardiac puncture was performed on euthanized animals to collect blood in EDTA-containing (plasma) and non-heparinized (serum) vacutainer tubes for hematological (CBC, HbA1c) and biochemical (Urea, creatinine, and LFT) analysis, respectively.
Oral glucose tolerance test (OGTT)The experimental rats were divided into 7 groups each having 3 males and 3 females and they were fasted for 12 hours (for food). The animals were divided into control (glucose 2 g/kg), negative (glucose 2 g/kg + DMSO1mL/kg), and positive control (glucose 2 g/kg + standard drug: Gliclazide 200 mg/kg), and treatment groups divided on the basis of doses of FO and its fractions mentioned below).
Five doses (50, 100, 200,400, and 600 mg/kg) of each of C.anthelminticum FO and its fraction (HF, CF, and EF) were orally administered to their respective groups followed by a glucose load of 2 g/kg. Blood from the tail veins of rats was used to evaluate glucose levels at various time intervals (0, 30, 60, and 120 minutes) using a glucometer (ACCU-CHEK Roche, Switzerland) [38]. Upon completion of the OGTT study percent glycemic change between the control and test, groups were calculated [39].
AnimalsMale Albino Wistar rats (n = 65, body weight = 180-280 ± 20 g) were procured from DUHS (Dow University of Health Sciences, Karachi). Polycarbonate cages were used to house the rats individually; they were acclimated to 12 hours of light and dark cycle for a week at a 22 ± 3 °C temperature and 50 ± 10% humidity. During the acclimatization period and before dietary intervention rats had free access to sterilized water and a standard rat diet. The Ethical Review Board for Animal Research and Ethics, Dow University of Health Sciences approved the study (AR.IRB-21/DUHS/Approval/2021/037).
Induction of diabetes in male Wistar ratsMale Wistar rats (10-12 weeks old; body weight 180-230 g) were separated into 6 treatment groups (n = 10 each) and a control group (n = 5). According to the groupings, the rats in the control group were fed with a normal diet whereas the ones in the other six treatment groups were fed a high-fat high-fructose (HF-HFr) diet for 28 days (i.e. 4 weeks). The HF-HFr diet used in this study mentioned in (Table 1) was the modification of protocol described by Yoo S et.al [40]. At the end of the 28th day, a single dose of STZ (60 mg/kg) in a citrate buffer (0.1 M, pH 4.5) was injected intraperitoneally into the 12-hour fasted rats in the treatment groups to develop T2DM. On the third day (i.e.72 hours) after STZ injection, FBG levels were measured from the tail vein of each rat using a glucometer, and rats having (FBG) levels of 230 mg/dL and above were considered as diabetic and randomly divided into 6 treatment groups (Fig. 1A).
Table 1 Composition and ingredients of experimental diet. RD: rats received a regular diet and 30 Frc + 45 Fat: rats received a 45 kcal% fat with a 30% fructose dietExperimental designSixty-five male Wistar rats were divided into seven groups: 10 animals in each group, except the normal control group (5 animals). The C. anthelminticum FO and its fractions were administered orally at a dose of 200 mg/kg to treatment groups from day 31-63 (i.e. 4 weeks). The dosage for C. anthelminticum oil was calculated on the basis of the acute toxicity study and oral glucose tolerance test results. The treatment was given daily for 4 weeks. During the study, body weight and FBG were measured weekly using a weighing machine and ACCU-CHEK glucometer, respectively.
Group 1(NC) - Normal control rats; normal diet and treated with distilled water (1 mL/kg).
Group 2 (DM Control) - Diabetic control rats; rats were fed HF-HFr diet and were administered 60 mg/kg of STZ and orally administered 0.01% DMSO (1 mL/kg).
Group 3 (DM Glic); Diabetic rats (fed HF-HFr diet and administered 60 mg/kg of STZ) were treated with the reference drug; Gliclazide (200 mg/kg).
Group 4 (FO); rats with DM (fed HF-HFr diet and administered 60 mg/kg of STZ) were treated with FO (200 mg/kg) of C. anthelminticum seeds.
Group 5 (HF); rats with DM (fed HF-HFr diet and administered 60 mg/kg of STZ) were treated with HF (200 mg/kg) of C. anthelminticum seed oil.
Group 6 (CF); rats with DM (fed HF-HFr diet and administered 60 mg/kg of STZ) were treated with CF (200 mg/kg) of C. anthelminticum seed oil.
Group 7 (EF); rats with DM (fed HF-HFr diet and administered 60 mg/kg of STZ) were treated with EF (200 mg/kg) of C. anthelminticum seed oil.
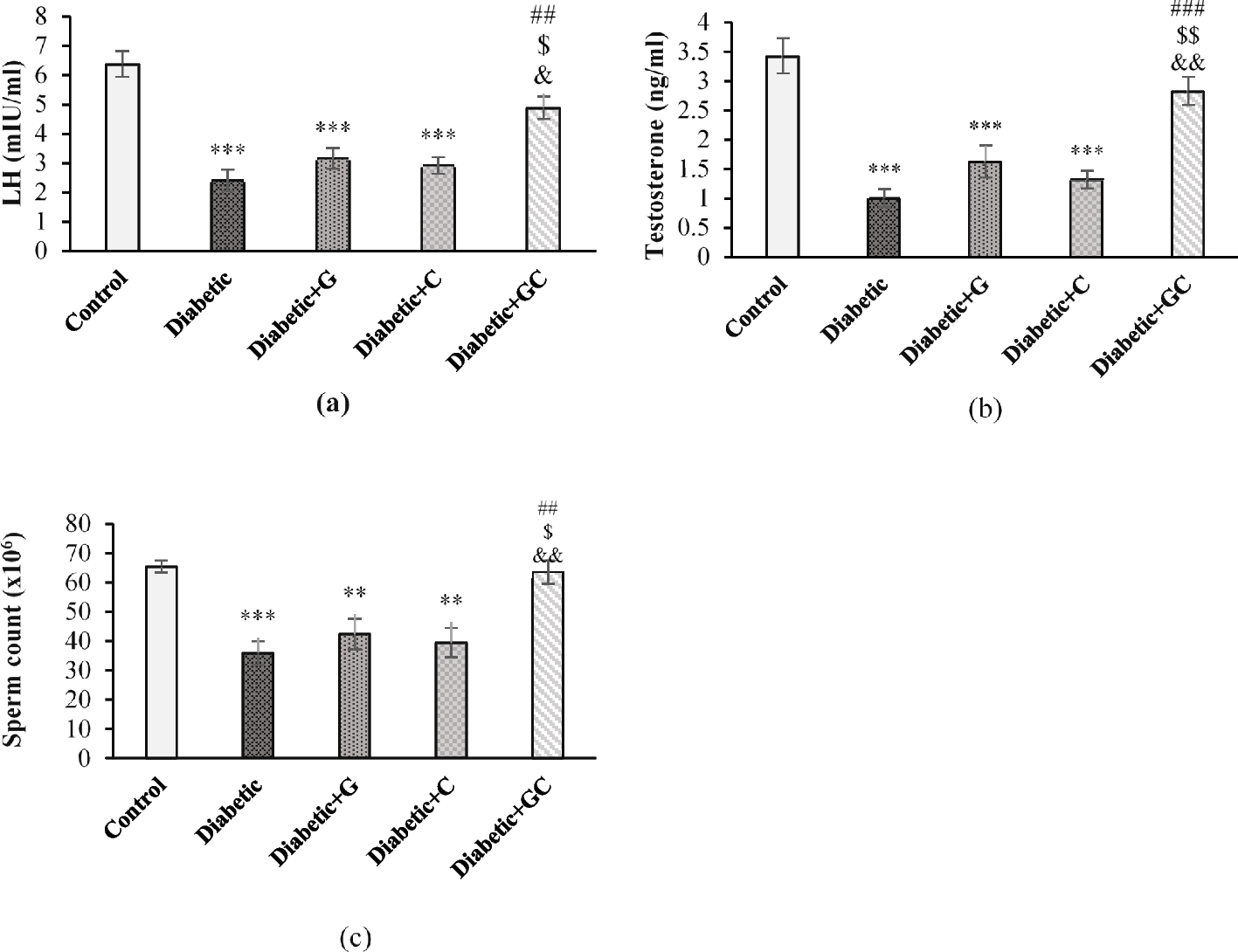
Biochemical analysisOn the 63rd day, the animals were sacrificed by intraperitoneal injection of Xylazine 7 mg/kg and Ketamine 60 mg/kg [37]. The blood samples were collected by cardiac puncture and centrifuged at 2000 x g for 15 minutes to separate serum for biochemical analysis. Serum insulin concentrations were determined according to the manufacturer’s instructions using a rat enzyme-linked immunoassay (ELISA) test kit (Bioassay Technology Laboratory Insulin ELISA kit Catalog no e0707RA). Glycated hemoglobin (HbA1c) and renal function assessment biomarkers such as serum creatinine and urea (mg/dL) were evaluated using commercially available spectrophotometric assay kits (Atellica Solutions, Siemens Healthcare). Homeostatic Model Assessment of Insulin Resistance (HOMA-IR), pancreatic β-cell function (HOMA β) and insulin sensitivity were calculated using the formulae: HOMA-IR = fasting insulin (μU/mL) × fasting glucose (mmol/L)/22.5 [41], HOMA β-cell (20 x insulin U/L/blood glucose - 3.5) and Insulin sensitivity = 1/log (fasting insulin U/L) x log (Fasting glucose mg/dl), respectively [41, 42]. The kidney tissues were excised, washed with ice-cold saline, and preserved in formalin 10% and phosphate buffer saline (PBS) for histopathological and PCR analysis, respectively.
Histopathology of renal tissuesAfter animals were sacrificed, the collected renal tissue was harvested, sectioned longitudinally, and fixed with 10% neutral buffer formalin for 48 hours. Followed by dehydration with gradient alcohol and transparentize with xylene, waxed, embedded, and sectioned. The 3 to 4 μm thick sections were Hematoxylin-Eosin (H&E) stained for general morphological analysis. The pathological changes in the kidney were observed under a compound microscope [43].
Homogenate preparation of renal tissueAll the tissues excised from both the control and experimental rats were placed in PBS and kept at − 80 °C. For homogenization, a 100 mM phosphate buffer with neutral pH was used. After complete tissue homogenization, the clear solution was centrifuged at 10,000×g for 15 minutes in order to remove any debris. The collected supernatant was used for further experimentation.
Determination of renal lipid peroxidation (LPO)The reagent TBA:HCl:TCA (15%:0.2 N:0.37%) was mixed with the kidney homogenate with a ratio of 1:1:1 (v/v). The mixture was then heated in boiling water for 15 minutes and was brought to room temperature for centrifugation at 5000 x g for 5 minutes. The absorbance was taken at 553 nm along with blank and the percent inhibition was calculated [44].
Determination of renal superoxide dismutase (SOD) activityThe enzymatic activity of the superoxide dismutase (SOD) was determined by Misra and Fridovich, 1972 [45]. The prepared homogenate was mixed with 0.3 mM of freshly prepared epinephrine and 0.05 M carbonate buffer (pH 10.2). The absorbance was calculated at 480 nm every 30s for 150 s. The 50% inhibition of the rate of autoxidation of epinephrine measured as a change in absorbance /min was employed in calculating one unit of enzyme activity.
Determination of renal catalase (CAT) activityThe catalase activity in the supernatant of kidney homogenate was assayed spectrophotometrically at 620 nm as described by Sinha [46]. The reaction mixture (1.5 mL) consisted of 0.1 mL of supernatant of kidney tissue homogenate, 0.4 mL of 2 M H2O2, and 1.0 mL of 0.01 M pH 7.0 phosphate buffer. The 2 mL of dichromate-acetic acid reagent (5% potassium dichromate and glacial acetic acid were mixed in a 1:3 ratio) was added to the solution to stop the reaction and the absorbance was measured.
Determination of renal HMG-CoA reductase activityThe activity of HMG-CoA reductase was determined in terms of the HMG-CoA/mevalonate ratio in kidney homogenate. The kidney homogenate was prepared in sodium arsenate solution. The homogenate was taken with an equal volume of dilute perchloric acid (PCA) mixed and incubated for 5 minutes at room temperature followed by a centrifuge at 3000 rpm for 10 minutes. 1.0 mL of kidney supernatant was collected in each of the two test tubes and allowed to react with 1.5 mL of ferric chloride and 0.5 mL of 2 M hydroxylamine reagent (alkaline pH = 5.5 in case of HMG-CoA and acidic pH = 2.1 in case of mevalonate) and incubated for 10 min. Absorbance was determined at 540 nm followed by a calculation of the HMG-CoA/mevalonate ratio [47].
Determination of renal reduced glutathione (GSH) level and glutathione peroxidase (GPx) activityThe GSH levels in the kidney homogenate was determined by using the procedure of Ellman (1959). Kidney homogenate (1.0 mL) was mixed with 0.1 mL of 25% TCA and the precipitate was removed by centrifuge at 5000 x g for 10 min. 0.1 mL of supernatant was removed and added to 2 mL of 0.6 mM DTNB (5,5′-dithiobis nitrobenzoic acid) prepared in 0.2 M sodium phosphate buffer (pH 8.0). The absorbance was read at 412 nm [48].
GPx activity was measured by the method described by Rotruck et al, 1973. The reaction mixture contained 0.2 mL of 0.4 M Tris-HCl buffer pH 7.0, 0.1 mL of 10 mM sodium azide, 0.2 mL of tissue homogenate (homogenized in 0.4 M, Tris-HCl buffer, pH 7.0), 0.2 mL glutathione, and 0.1 mL of 0.2 mM hydrogen peroxide. The contents of the mixture were incubated at 37 °C for 10 min. The reaction was arrested by 0.4 mL of 10% TCA and centrifuged. The supernatant was assayed for glutathione content by using Ellman’s reagent (19.8 mg of 5,5′-dithiobis nitrobenzoic acid (DTNB) in 100 mL of 0.1% sodium nitrate) [49].
Determination of levels of NF-κB p65 DNA binding activityThe ELISA of transcription factor NF-κB was carried out on the renal tissues homogenates as per manufacturer instruction (USCN Catalog no. SEB824Ra). The optical density of protein-bound NF-κB was measured at 450 nm.
Reverse transcription quantitative real-time PCR (RT-qPCR) analysisThe kidneys of dissected animals were stored in PBS solution at − 80°C to preserve the RNA integrity. Total RNA was extracted using the TRIzol® Reagent. The integrity of the RNA was checked on 1% agarose gel electrophoresis. The quantitation of RNA was done with a nanodrop. Afterward, the complementary DNA (cDNA) was synthesized using a Thermo Scientific RevertAid First Strand cDNA Synthesis Kit (Catalog no. K1691 Thermofisher Scientific, USA). The PCR cDNA with eight different sets of primers was carried out using SYBR™ Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) and was performed in StepOnePlus Real-time PCR system. Table 2 contains the primer sets used in RT-qPCR. The gene HPRT-1 was used as a reference gene to measure the relative expression of the mRNA in a sample. The amplification PCR program included 1 cycle of 94 °C for 10 min, followed by 35 cycles of 94 °C for 1 min, 60 °C for 40 sec, and 72 °C for 30 sec, and a final elongation cycle at 72 °C for 5 min using an Applied Biosystems, Foster City, CA, USA. Each sample was run in duplicates in order to ensure the reproducibility of the reaction [32].
Table 2 Forward and reverse primers (5′ → 3′) for reverse transcriptase-real time PCR (RT-qPCR)Statistical analysisThe results are expressed as the mean ± standard error mean (S.E.M). Statistical analysis was carried out using the One-way ANOVA followed by least significant difference (LSD) multiple comparisons post-hoc test. A value of p < 0.05 was considered statistically significant. IBM SPSS v. 26 software (Chicago, IL, USA) was used for statistical analysis.
留言 (0)