
記住我
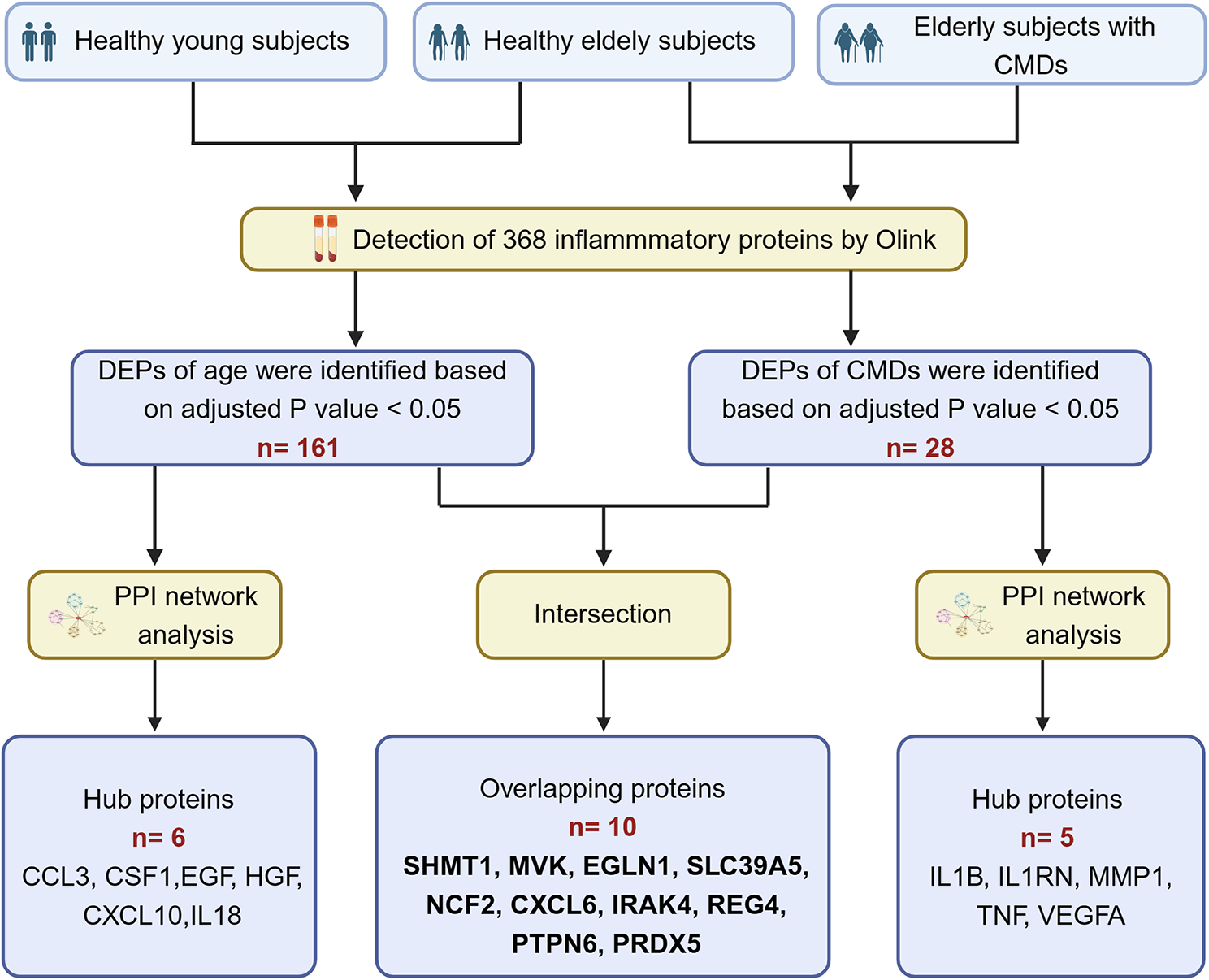
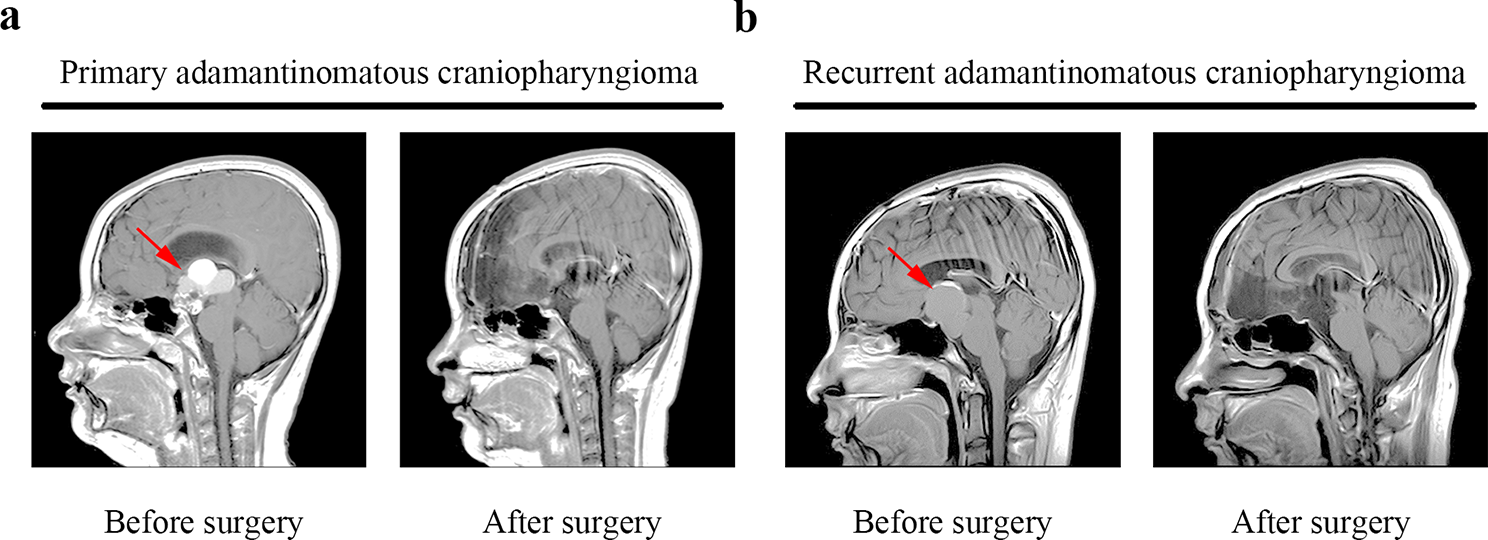

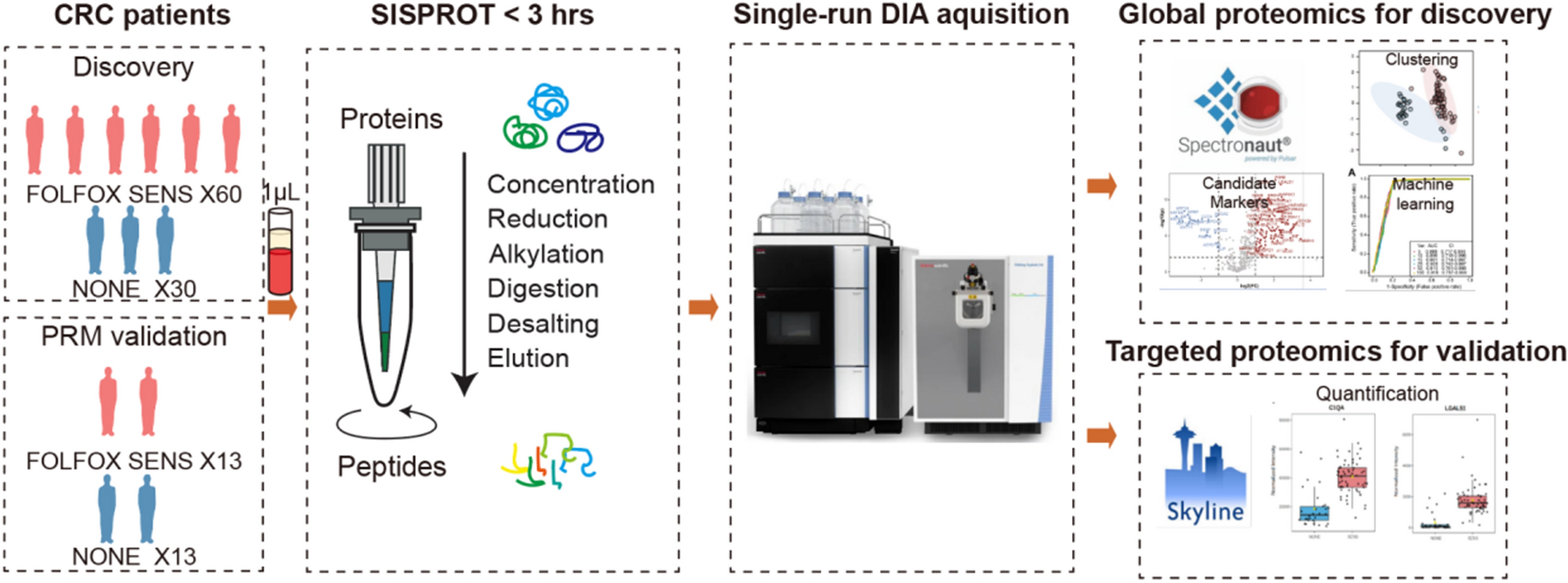
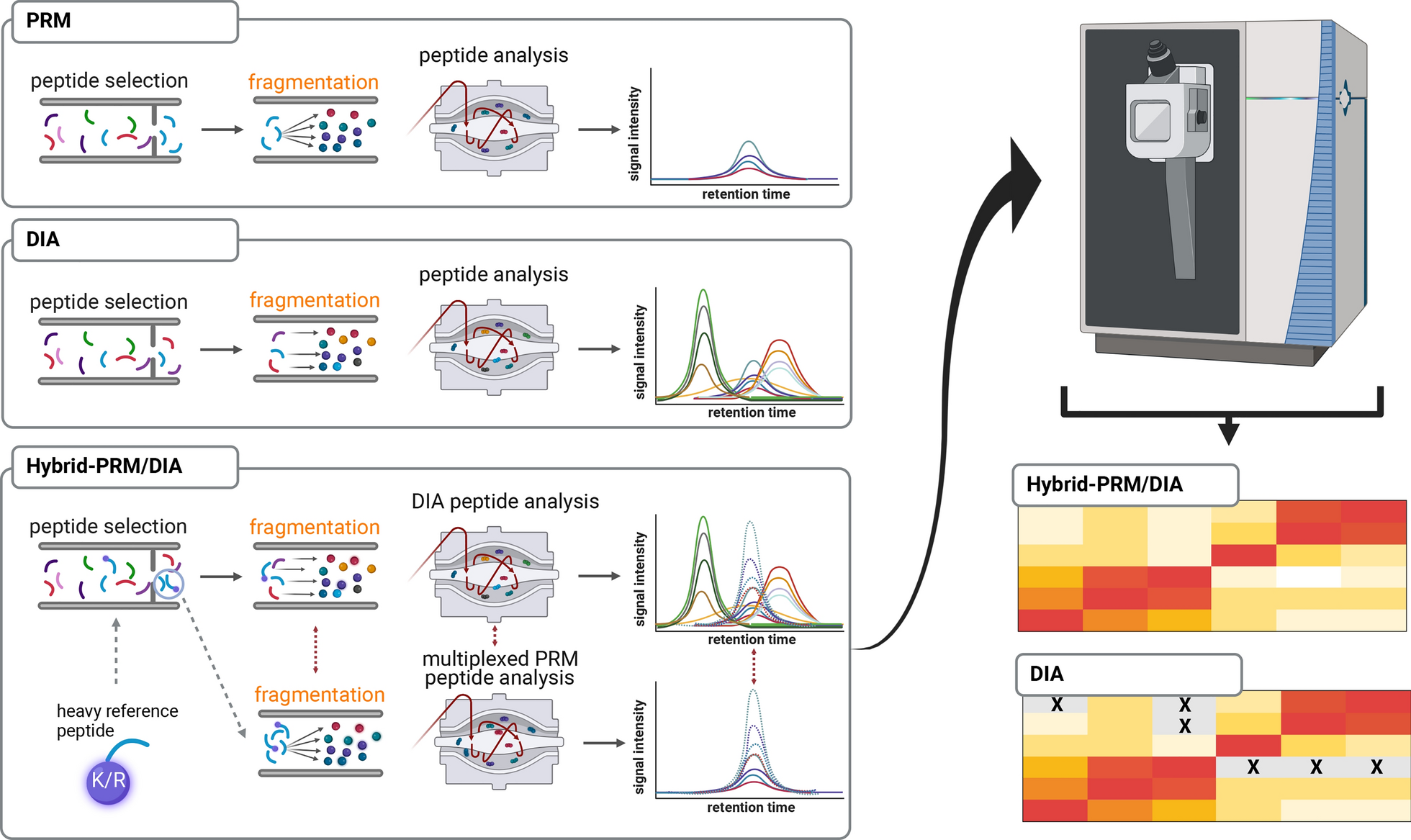


This study contained two groups, including 20 LUAD patients and 20 healthy controls (NL). The average age was 57.8 ± 10.8 and 45.1 ± 11.4 in LUAD and NL groups, and LUAD included 6 male and 14 female, while NL contained 7 male and 13 female. All LUAD patients were classified as stage I according to 7th version, and 7 had smoke history while 13 were never smoking. All clinical features of all participants were listed in Table 1.
Table 1 The clinical features of all participates in this studyTo explore N-glycoprotein sites for lung cancer diagnosis, plasma were obtained from LUAD and NL controls and were extracted and labelled. After fractionation and enrichment, LC–MS/MS was performed to investigate the N-glycoprotein levels in all samples. The differential N-glycoprotein levels between LUAD and NL were analyzed based on databases. And the candidate N-glycoprotein sites were combined to study the role of novel biomarkers for future lung cancer diagnosis. The study procedures were listed in Fig. 1.
Fig. 1
Workflow of N-glycosylation analysis in LUAD patients and NL controls. Trypsin was added into all samples for protein digestion and then processed by TMT kit/iTRAQ kit. High pH reverse-phase HPLC was performed to fractionate tryptic peptides and then dissolved in NETN buffer for enrichment. The peptides were then subjected to tandem mass spectrometry (LC–MS/MS) in Q ExactiveTM Plus. A data-dependent procedure was then conducted to peptides and alternated between one MS scan followed by 20 LC–MS/MS scans with 15.0 s dynamic exclusion
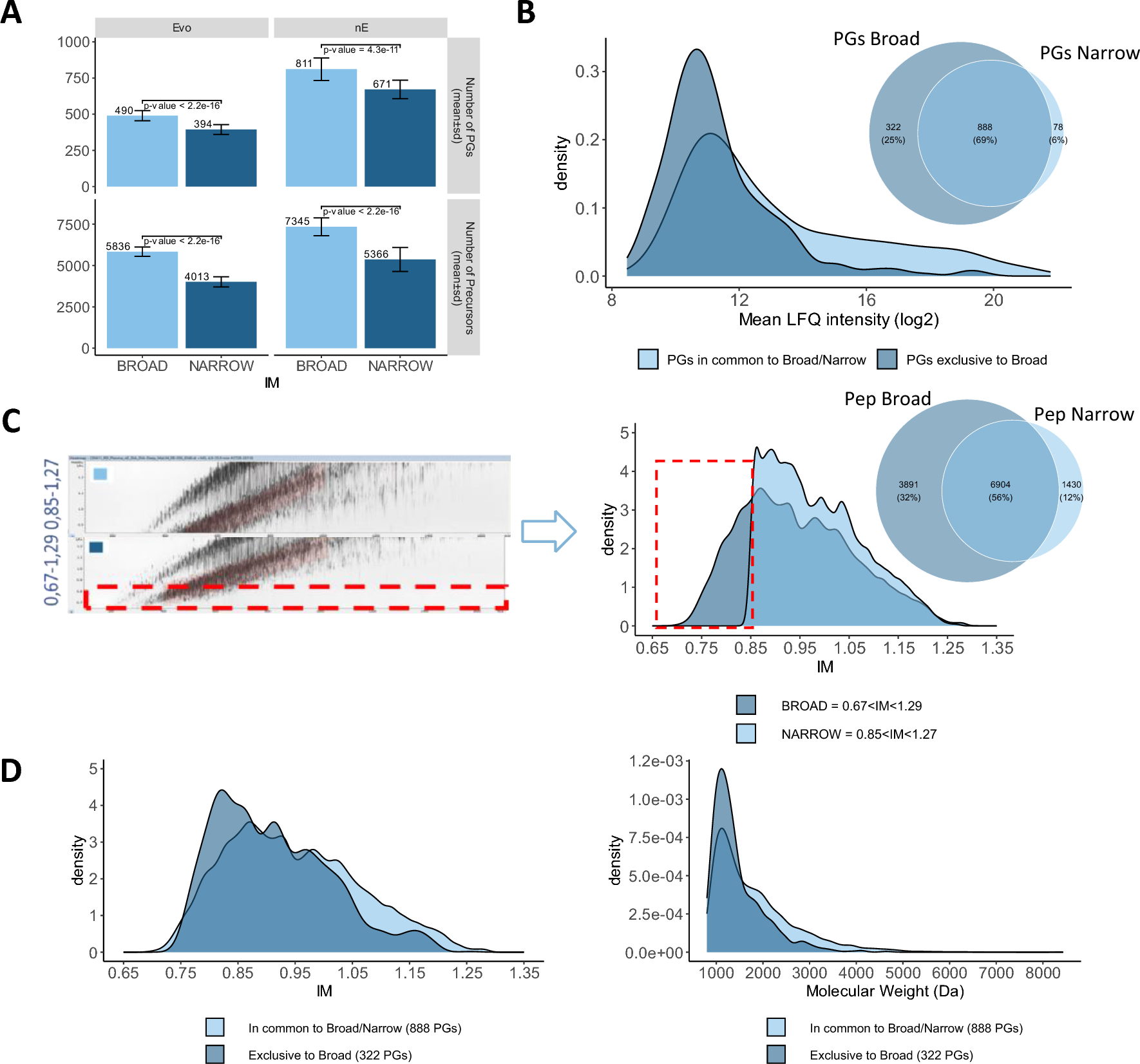
Characteristics of the identified N-glycoproteinsNext, we performed enriched peptides and detected by LC–MS/MS to identify N-glycoproteins in obtained plasma samples. In comparison to NL samples, we obtained total 383,675 spectrums in LUAD patients and 18,566 matched spectrums were identified based on protein datasets (Additional file 1: Table S1). We then identified total 4385 peptides in matched spectrums. In all peptides, 1399 were belongs to modified peptides which contained 502 identified sites. In LUAD samples, 478 sites were quantified which were identified from 275 proteins (Fig. 2A).
Fig. 2
Characteristics of identified N-glycoproteins. A Total number of identified N-glycosylation proteins. B The number of modification sites per protein. C Pie chart showed the number and proportions of single and multiple N-glycosylated sites in LUAD. D Overlap numbers of glycoproteins in LUAD samples. E Overlap numbers of glycosylation sites in LUAD samples
In this study we identified total 478 sites in 263 proteins in LUAD patients. The number of N-glycosylated sites assigned to all proteins ranged from 1 to 11 with average degree of glycosylation was 2.5. More than half of glycoproteins (270/478, 57.1%) carried only a single N-glycoprotein site, 110 (23.6%) of them harbored double N-glycoprotein sites (Fig. 2B, C). Triple and four N-glycoprotein sites were 41 (9.1%) and 27 (6.2%), respectively. And the rest 17 (4%) contained five (8, 2.5%), eight (1, 0.4%), nine (3, 0.7%) and eleven (1, 0.4%) sites (Fig. 2B, C). And our results also indicated the overlap numbers in all samples in both protein and N-glycoprotein sites (Fig. 2D, E). In summary, multiple proteins and N-glycosylation sites were identified in LUAD patients, which could be conducted to further analysis to identify candidate biomarkers for future clinical application.
Disease-associated changes in N-glycopeptide abundance in LUADIn comparison to NL samples, 39 differential N-glycosylation sites were obtained in LUAD. In all differential sites, 17 increased in LUAD patients, such as APOB-2982, SERPINC1-224 and APOB-1523, while 22 decreased in lung cancer samples, including ITGB3-125 and VWF-235 (Fig. 3A). We analyzed the cellular distribution of the differential proteins, and the result indicated 24 proteins were extracellular, 3 proteins were endoplasmic reticulum, while the cytoplasm, cytoskeletio, mitochondria and plasma membrane contained 1 protein (Fig. 3B).
Fig. 3
Disease-associated variations in N-glycopeptide abundance in LAUD. A Volcano revealed increased and decreased N-glycosylation sites in LUAD and NL samples. B Classification of identified N-glycosylation proteins based on subcellular loction. C Sequence motifs located nearby the target asparagine in enriched glycosylation sites. D Heatmap showing the relative frequency of amino acids in the proximity of asparaine (enrichment, red; depletion, green)
The neighborhood residues of glycosylated asparagines could determine the specificity of LUAD. MoMo was conducted to obtain the characteristic sequence of modified sites and their enrichment statistics. As shown in Fig. 3C, 2 conserved amino acids flanking the glycosylated asparagine residues (from − 10 to + 10) were defined. These motifs included N-x-T-*-Y and N-x-S, where x represented any amino acid except proline and the asterisk denoted a random amino acid. Based on analysis of hierarchical clusters, threonine and serine displayed the highest probability at the position + 2, while the frequency of a proline residue in the proximity was markedly underrepresented (Fig. 3D). Taken together, our results suggested a preference motif exposed to the surface of glycoproteins.
Analysis and annotation of differentially N-glycosylated proteins in LUADTo elucidate the potential functions of those quantifiable proteins in LUAD samples, we analyzed the quantifiable proteome data set for three enrichment gene ontology (GO) categories: molecular function, cellular compartment and biological process. Based on accumulative normal distribution, we divided all pathways into 4 quantiles: Q1 (< 0.769 fold change), Q2 (0.769–0.833 fold change), Q3 (1.2–1.3 fold change) and Q4 (> 1.3 fold change). In the biological process category, the significant increased pathways enriched in cell migration process such as tissue remodeling, cell growth, cell–matrix adhesion and actin cytoskeleton organization (Q1), and also contained immune-related regulation including leuckocyte chemotaxis, lymphocyte migration, cytokine stimulus and T cell migration (Q2). In Q3 and Q4 analysis, we also found that activated pathways in LUAD samples aggregated in innate immune response, endocytosis (Q3) and in metabolic transport process including protein, lipid, sterol and cholesterol (Q4) (Fig. 4A). As to cellular component analysis, the results revealed that assembled components in LUAD patients were intrinsic, integral, vesicle, cytoplasmic and organelle membrane (Q1), trans-Golgi network, vesicle transport and secretory granule lumen (Q2), metabolic components (protein-lipid complex, plasma lipoprotein particle and lipoprotein particles) (Q3) and endocytic vesicles (Q4) (Fig. 4B). Finally we analyzed molecular function pathways in LUAD samples, we observed enrichment focused on binding activity, including protease, fibronectin, carbohydrate and sulfur compound (Q1), enzyme, cytokine and growth factor (Q2), as well as transport activity (Q3) and lipoprotein receptor binding (Q4) (Fig. 4C). We then analyzed KEGG pathways in LUAD samples and found that the enrichment contained neutrophil extracellular formation, platelet activation (Q1) and amoebiasis (Q2) (Fig. 4D).
Fig. 4
Annotation of differential N-glycosylated proteins in LUAD samples. A–D. Functional enrichment-based clustering analysis for quantified glycoproteome. A biological process analysis, B cellular component, C molecular function analysis, D KEGG pathway analysis. E PPI network analysis. Each node represents an N-glycoprotein and each edge represents the interaction between proteins
Finally STRING protein–protein interaction (PPI) dataset was performed to analyze the protein–protein interactions in identified proteins. The results indicated that APOB, SERPINC1 and CLU were the core nodes in interaction network, which interacted with multiple proteins such as IGF2R, C8A, HPX, HRG and ADAM10. FN1 and SERPING1 also acted as central connection nodes, which interacted with SERPINA5, ITGB3, VWF and F5 (Fig. 4E). Taken together, our results indicated that by TMT labeling and LC–MS/MS sequencing, we identified multiple differential proteins and N-glycosylation sites in LUAD patients, which harbored multiple pathways in tumor-related abnormal metabolism and protein transport.
The role of N-glycosylation sites in diagnosis of LUADTo further explore the diagnostic accuracy of candidate biomarkers in LUAD, we performed ROC analysis to define the sensitivity (SN) and specificity (SP) of identified N-glycosylation sites. The results suggested that multiple N-glycosylation sites in proteins harbored valuable roles in lung cancer diagnosis. The tope 4 proteins were ITGB3-680, APOB-1523, APOB-2982 and LPAL2-101, which all showed AUC (area under curve) > 80.0% (Fig. 5A). The most important site was ITGB3-680, the AUC was 99.2%, SN (sensitivity) and SP (specificity) were both 95.0% in compared with NL group. In APOB-1523 analysis, the AUC was 89.0%, SN and SP were 70.0% and 95/0%, respectively. In APOB-2982 analysis, AUC was 86.8%, SN was 45.0% and SP was 95.0% when compared with NL group. The AUC in LPAL2-101 analysis was 81.1%, while SN was 47.4%, SP was 95.0% (Fig. 5A).
Fig. 5
The concentration and ROC analysis of differential N-glycosylation sites identified in LUAD patients. A The concentration and ROC analysis of N-glycosylation sites with AUC > 80.0%. B, C The concentration and ROC analysis of N-glycosylation sites with AUC > 70.0%. **p < 0.01, ***p < 0.001
Besides top 4 proteins, we also analyzed other candidate proteins. We found that AUC of CLU-291 was 78.5%, SN was 50.0% and SP was 95.0%. And the AUC, SN, SP of VWF-2357 were 76.8%, 50.0% and 95.0%, respectively. In C8A-437 analysis, the AUC was 76.5%, SN was 45.0%, SP was 95.0%. In CD109-247 analysis, we obtained AUC was 76.5%, SN was 25.0% and SP was 95.0% (Fig. 5B).
Finally we investigate ECM1-444, CFH-882, VNN1-283 and AFM-33. The analysis results indicated that in ECM1-444, the AUC was 75.6%, SN was 30.0% and SP was 95.0%. In CFH-882, the AUC was 75.5%, SN was 25.0%, SP was 95.0%. In VNN1-283 analysis, the AUC was 75.5%, SN was 60.0% and SP was 90.0%. In AFM-33 analysis, the result showed that AUC was 75.3%, while SN and SP were 60.0% and 95.0%, respectively (Fig. 5C).
In summary, our study revealed multiple N-glycosylation sites harbored highly potential diagnostic value in LUAD diagnosis.
Combination analysis of novel N-glycosylation sites by machine learning modelIN this part, we introduced machine learning model to test the diagnosis efficiency of candidate biomarkers in lung cancer. We divided all participants into training set (establish model and adjust parameters, 16 cases) and test set (evaluate the model, 4 cases). By combining feature selection, machine learning algorithm, classifier integration method and dataset validation, random forest model was conducted to determine whether the proteomic profile had cancer-specific features for lung cancer diagnosis (Fig. 6A). Due to small sample sizes of two data sets, 2/3 individuals in the training set were selected to grow decision trees by boostrapping and the remaining participants were used as out of bag samples for cross-validation importance. In feature selection, each sample was represented by feature vector, which contained 24 expression features and each expression feature has different ability to distinguish different types of samples. Univariate feature analysis was introduced to quantify the ability of expression features in distinguishing different samples and we could calculate the correlation between each feature and sample types by variance test. Based on this method, the feature scores and p value of candidate molecules revealed the top 15 molecules in candidate N-glycosylation sites, including ITGB3-680, APOB-2982, CLU-291, ECM1-444, C8A-437, VNN1-283, LPAL2-101, APOB-1523, BTD-56, AFM-33, APOB-3411, AFM-402, CFH-882, CRISP3-270, SERPINA5-262, IGHG4-177 and SERPING1-238, which were performed for model construction (Fig. 6B). Next, we analyzed the Pearson correlation coefficient to understand the linear correlation of top 5 proteins (ITGB3-680, APOB-2982, CLU-291, ECM1-444 and C8A-437), the results indicated that ITGB3-680, ECM1-444 and C8A-437 correlated closely (0.459 and 0.368) while APOB-2982 and CLU-291 had correlation (0.357) (Fig. 6C). Finally machine learning model was established to evaluate the role of candidate biomarkers in lung cancer diagnosis. In this study we used logistic regression, support vector machine and random forest as base classifier to construct voting classifier. To evaluate the differences between prediction and actual category, four calculation accuracy index, including sensitivity, specificity, Mattthews’ correlation coefficient and AUC (area under curve) were introduced in this model. In all expression features, the optimal expression feature subset in current data set was selected to obtain optimal prediction accuracy by incremental feature selection (IFS). The AUC curves in training and test set were obtained by plotting the true positive rate against the false positive rate under different cut-off values, and the result indicated that AUC reached 100% in both training and test sets (Fig. 6D). In summary, the machine learning model revealed that combination of N-glycosylation sites had important application in lung cancer diagnosis.
Fig. 6
Combination analysis of candidate biomarkers by machine learning. A Analysis schema of machine learning. B Feature score of candidate N-glycosylation sites identified by feature selection. C Pearson correlation coefficient in Top 5 N-glycosylation sites in feature score. D AUC in training (16 cases) and test set (4 cases) by plotting the true positive rate against the false positive rate under different cut-off values
留言 (0)