Animals
Six-week-old male DBA/1J and C57BL/6 mice were purchased from Orient Bio Inc. (Seongnam, Korea). The mice were maintained under specific pathogen-free conditions at the Institute of Medical Science of the Catholic University of Korea with ad libitum access to water and were fed standard mouse chow (Ralston Purina, St. Louis, MO, USA). All experimental procedures were performed in accordance with the Laboratory Animals Welfare Act, the Guide for the Care and Use of Laboratory Animals, and the Guidelines and Policies for Rodent Experiments provided by the Department of Laboratory Animals of the Institutional Animal Care and Use Committee of the Catholic University of Korea and conformed to all National Institutes of Health (USA) guidelines (permit numbers: 2016-03-02, 2017-08-03, 2018-05-28). To minimize animal suffering, all mice were fully anesthetized under gas anesthesia using isoflurane (2–2.5%) and sacrificed by cervical dislocation.
CIA induction and treatment with EC-18
CIA was induced as described previously [26]. To induce CIA in mice, type II collagen (CII) was dissolved overnight in 0.1 N acetic acid (4 mg/mL) with gentle rotation at 4°C. Seven-week-old male DBA/1J mice were injected intradermally at the base of the tail with 100 μg of CII emulsified in Freund’s adjuvant (Chondrex). Two weeks later, 100 μg of type II collagen dissolved and emulsified 1:1 with incomplete Freund’s adjuvant (Difco) was administered to the hind leg of mice as a booster injection. To examine the preventive effects of EC-18 on the development of arthritis, mice were administered via oral gavage with the vehicle only (2.5 % DMSO in PBS), baricitinib (3 mg/kg, 0.4 % DMSO in PBS), tofacitinib (15 mg/kg, 1.2 % DMSO in PBS), or EC-18 (250 mg/kg, 2.5 % DMSO in PBS), which was developed as an oral treatment, once daily from day 21 after the first immunization. To evaluate the synergistic effects of EC-18 with MTX, mice were administered via oral gavage with the vehicle only (2.5 % DMSO in PBS), MTX (1.5 mg/kg, 0.1 % DMSO in PBS), EC-18 (125 mg/kg, 2.5 % DMSO in PBS), or MTX combined with EC-18 once daily from day 21 after the first immunization. The mice were scored using the arthritis index, which indicates the onset and severity of joint inflammation, twice weekly for up to 8–10 weeks after the first immunization.
Clinical assessment of arthritis
To assess the severity of joint inflammation, the arthritis index was scored twice weekly from the onset of arthritis for up to 8–10 weeks after the primary immunization. The severity of arthritis was assessed on a scale of 0–4 on 4 paws per mouse (maximum score of 16) according to the following criteria, as described previously [27]: 0 = no edema or swelling, 1 = slight edema and erythema limited to the foot or ankle, 2 = slight edema and erythema from the ankle to the tarsal bone, 3 = moderate edema and erythema from the ankle to the tarsal bone, and 4 = edema and erythema from the ankle to the entire leg. Incidence was calculated as 25% of the presence of arthritis symptoms in one foot. The arthritic score and incidence for each mouse were expressed as the sum of the scores of four limbs.
Antibodies
The following antibodies were used for immunohistochemistry: rabbit polyclonal anti-IL-6 (1:50, ab7737), rabbit polyclonal anti-TNF-α (1:150, ab6671), rabbit polyclonal anti-IL-17 (1:600, ab79056), rabbit monoclonal anti-STAT3 (phosphor Tyr705) (1:100, ab76315), rabbit monoclonal anti-tartrate-resistant acid phosphatase (TRAP) (1: 100, ab191406), and rabbit IgG, monoclonal [EPR25A] isotype control (1:100, ab172730) were from abcam; rabbit polyclonal anti-IL-1β Ab (1:400, NB600-633) was from Novus Biologicals. The following antibodies were used for western blot: mouse monoclonal anti-β-actin Ab (1:1000, sc-47778) were from Santa Cruz Biotechnology; rabbit monoclonal anti-STAT3 (phosphor Y705) (1:2000, #9145), rabbit polyclonal anti-STAT3 (phosphor S727) (1:1000, #9134), and mouse monoclonal STAT3 (124H6) Ab (1:1000, #9139) were from Cell signaling; rabbit monoclonal anti-GAPDH [EPR16891] (1:2000, ab181602) was from abcam. The following antibodies were used for flow cytometry for mouse: rat monoclonal PerCP-Cyanine5.5 anti-CD4 (0.25 μg/test, #45-0042-82) and rat monoclonal FITC anti-IL-17A Ab (0.25 μg/test, #11-7177-81) were from Thermo Fisher Scientific; rat PE/Cyanine5 IgG2a κ isotype control (#400510) and rat FITC IgG2a, κ isotype control Ab (#400505) were from BioLegend. The following antibodies were used for flow cytometry for human: mouse monoclonal PE/Cyanine7 anti-human CD4 (5 μl per test, #300512) and rat monoclonal FITC IgG2a, κ isotype control Ab (#400505) were from BioLegend; mouse monoclonal PE anti-human IL-17 (0.25 μg/test, #12-7179-42), mouse monoclonal APC anti-CD25 (0.125 μg/test, #17-0259-42), rat monoclonal FITC anti-Foxp3 (0.5 μg/test, #11-4776-42), mouse PE-Cyanine7 IgG1, κ isotype control (# 25-4714-42), mouse PE IgG1, κ isotype control (#12-4714-42), and mouse APC IgG1, and κ isotype control Ab (#17-4714-42) were from Thermo Fisher Scientific.
Histopathology
Mouse joint tissues were fixed in 10% neutral-buffered formalin, decalcified in decalcifying agent (National Diagnostics, Atlanta, GA, USA), embedded in paraffin, and cut into sections 5 μm thick. The sections of ankle/tarsal bones were stained with hematoxylin and eosin (H&E) and safranin O and scored as described previously [28]. Inflammation was scored using the following criteria: 0, normal; 1, minimal infiltration of inflammatory cells and/or mild edema; 2, mild infiltration; 3, moderate infiltration; 4, marked infiltration; and 5, severe infiltration of the synovium by inflammatory cells. Bone erosion was scored using the following criteria: 0, normal; 1, clearly invisible bone resorption in trabecular and cortical bone; 2, clearly invisible bone resorption in slightly increased trabecular and cortical bone; 3, clearly visible bone resorption in the trabecular and cortical bones. Cortex thickness decreases and trabecular loss occurs; 4, no distortion on the cortical bone surface and the thickness of trabecular and cortical bone is reduced; 5, decreased cortical bone surface and thickness of trabecular and cortical bone. Cartilage damage was evaluated by staining with Safranin O and toluidine blue, and the extent of damage was scored using the following criteria: 0, normal; 1, minimal loss of cartilage without apparent chondrocyte loss or collagen disruption; 2, chondrocyte loss and/or collagen disruption and mild loss of cartilage (superficial); 3, moderate loss of chondrocyte loss and/or collagen disruption and cartilage (depth to middle zone); 4, marked loss of chondrocyte loss in various areas and/or collagen disruption and cartilage (depth to deep zone); and 5, severe chondrocyte loss and/or collagen disruption and (depth to tidemark) severe loss.
Immunohistochemistry
Sections were treated with 3% (v/v) H2O2 in methanol to block endogenous peroxidase activity. Immunohistochemistry was performed using the Envision Detection™ kit (DAKO Agilent Technologies Inc., Santa Clara, CA, USA). Tissue sections were incubated with primary antibodies against IL-6, TNF-α, IL-1β, IL-17, phosphorylated (p) STAT3 (Tyr705), and TRAP for 2 h at room temperature. Sections were then incubated with a biotinylated secondary Ab and streptavidin–peroxidase complex for 30 min. The final colored products were developed using chromogen diaminobenzidine, and the sections were examined under a photomicroscope (Olympus, Tokyo, Japan). Two independent, blinded observers assessed the number of positive cells per section at high-power field (magnifications ×400). To analyze osteoclast parameters, osteoclast surface normalized to the bone surface were assessed by bone histomorphometric analyses (ImageJ software).
Isolation of splenocytes and cell stimulation
Splenocytes and splenic CD4+ cells from normal C57BL/6 or CIA mice were isolated as described previously [29]. Mouse spleens were ground using sterilized glass slides with frosted ends and red blood cells were lysed in hypotonic ACK buffer (0.15 mM NH4Cl, 1 mM KCO3, and 0.1 mM EDTA, pH 7.4). The remaining splenocytes were filtered through a 40-μm cell strainer (Falcon, Durham, NC) and maintained in RPMI 1640 medium containing 5% fetal bovine serum (Thermo Fisher Scientific, MA, USA). To purify splenic CD4+ T cells, the splenocytes were incubated with CD4-coated magnetic beads and isolated on MACS separation columns (Miltenyi Biotec). Splenocytes were pretreated with EC-18 for 2 h and then stimulated with 100 ng/ml lipopolysaccharide (LPS) (Sigma-Aldrich #L4391) for 3 days. Th0 cells were stimulated only with 0.5 μg/ml anti-CD3 (BD Pharmingen #553057) and 1 μg/ml anti-CD28 (BD Pharmingen #553294) antibodies without the addition of cytokines. Th17 differentiation was induced via treatment with anti-CD3, anti-CD28, 20 ng/ml IL-6 (R&D systems #406-ML), 2 ng/ml transforming growth factor (TGF)-β (PeproTech #100-21C), 5 μg/ml anti-IFN-γ Ab (R&D systems #MAB485), and 5 μg/ml anti-IL-4 Ab (R&D systems #MAB404). For regulatory T (Treg) polarization, anti-CD3, anti-CD28, anti-IFN-γ anti-IL-4 antibodies, and TGF-β (5 ng/ml) were added to the culture. Splenic CD4+ cells were pretreated with EC-18 for 2 h and then stimulated under Th0, Th17, or Treg conditions for 3 days. To determine the levels of p- STAT3 by western blot, CD4+ cells were stimulated with IL-6 for 30 min or anti-CD3 and anti-CD28 Ab for 12 h in the presence of EC-18.
Isolation of human peripheral blood mononuclear cells and cell stimulation
Human blood was obtained from patients with RA (Table 1). The diagnosis of RA was confirmed according to the revised criteria of the American College of Rheumatology [30]. Normal healthy volunteers were included as controls. All patients provided informed consent in accordance with the guidelines of the Declaration of Helsinki. Approval by the ethics committee of Seoul St. Mary’s Hospital (Seoul, Korea) was obtained for all procedures (permit numbers: KC17MNSI0405, KC17TNSI0704). Peripheral blood mononuclear cells (PBMCs) were separated from the buffy coat using Ficoll–Hypaque (Pharmacia Biotech, Piscataway, NJ, USA). The cells were pretreated with EC-18 for 2 h and then stimulated with anti-CD3 Ab (0.5 μg/ml) for 12 h, 2 days, or 3 days.
Table 1 Clinical characteristics of patients with rheumatoid arthritisMouse in vitro osteoclastogenesis
Bone marrow-derived monocytes/macrophages (BMMs) were isolated from the tibias and femurs of the mice by flushing the bone marrow cavity with α-minimum essential medium (MEM; Invitrogen, Carlsbad, CA, USA). The cells were incubated with α-MEM containing 10% fetal bovine serum for 12 h to separate the floating and adherent cells. Floating cells (1 × 105 cells/500 μl) were cultured in the presence of M-CSF (10 ng/ml) for 3 days to form osteoclast precursor cells (preosteoclasts). Three days later, the nonadherent cells were washed out, and preosteoclasts were stimulated with EC-18 in the presence of 10 ng/ml macrophage colony-stimulating factor (M-CSF, PeproTech #300-25) and 50 ng/ml RANKL (PeproTech #310-01) for 4 days to generate osteoclasts. On day 2, the medium was replaced with fresh medium containing M-CSF, RANKL, and EC-18. TRAP staining (Cosmo Bio, #PMC-AK04F) was performed according to the manufacturer’s instructions. Mouse BMMs were prepared using the method described above and were cultured in 48-well OAAS plates (Osteogenic Core Technologies, Choongnam, Korea). Erosive areas were identified using the Tomoro analySIS TS Lite program (Olympus, Münster, Germany).
Human in vitro osteoclastogenesis
PBMCs (5 × 105 cells/500 μl) were incubated at 37°C for 2 h to separate nonadherent and adherent cells, and the adherent cells were then cultured with M-CSF (100 ng/ml) for 3 days. After 3 days, these preosteoclast cells were cultured further in the presence of M-CSF (25 ng/ml), RANKL (30 ng/ml), and EC-18 for 9 days to generate osteoclasts. On day 3, the medium was replaced with fresh medium containing M-CSF, RANKL, and EC-18. TRAP staining (Cosmo Bio) was performed according to the manufacturer’s instructions.
Analysis of gene expression using real-time polymerase chain reaction (PCR)
Total RNA was extracted using TRI Reagent (Molecular Research Center), and cDNA was synthesized with Dyne first-strand cDNA synthesis kit (Dyne Bio) according to the manufacturer’s protocol. Polymerase chain reaction amplification and analysis were performed using a LightCycler 2.0 instrument (Roche Diagnostics, Indianapolis, IN, USA) and the accompanying software (version 4.0). All reactions were performed using a SensiFAST™ SYBR® Hi-ROX kit (Meridian Bioscience, Cincinnati, OH, USA), according to the manufacturer’s instructions. The following primers were used for mouse sequences: TRAP, 5′-TCCTGGCTCAAAAAGCAGTT-3′ (sense), 5′-ACATAGCCCACACCGTTCTC-3′ (antisense); carbonic anhydrase II, 5′-TGGTTCACTGGAACACCAAA-3′ (sense), 5′-AGCAAGGGTCGAAGTTAGCA-3′ (antisense); calcitonin receptor, 5′-CGGACTTTGACACAGCAGAA-3′ (sense), 5′-AGCAGCAATCGACAAGGAGT-3′ (antisense); and β-actin, 5′-GTACGACCAGAGGCATACAGG-3′ (sense), 5′-GATGACGATATCGCTGCGCTG-3′ (antisense). The following primers were used for human samples: RORc, 5′-AGTCGGAAGGCAAGATCAGA-3′ (sense), 5′-CAAGAGAGGTTCTGGGCAAG-3′ (antisense); Foxp3, 5′-CACTGCCCCTAGTCATGGT-3′ (sense), 5′-GGAGGAGTGCCTGTAAGTGG-3′ (antisense); and β-actin, 5′-GGACTTCGAGCAAGAGATGG-3′ (sense), 5′-TGTGTTGGGGTACAGGTCTTTG-3′ (antisense). The levels of mRNA expression were normalized relative to that of β-actin mRNA.
Enzyme-linked immunosorbent assay (ELISA)
The levels of IL-6, IL-1β, TNF-α, IL-17, and IL-10 for mouse (DY406, DY401, DY410, DY421, DY417) or human (DY206, DY201, DY210, DY317, DY217B) were performed using DuoSet ELISA kits (R&D Systems). HRP–avidin (R&D Systems) was used for color development. The absorbance of each sample was measured at 405 nm (A405) using an ELISA microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Measurement of IgG2a and type II collagen-specific IgG2a
Blood was taken from the orbital sinus of mice, and the sera were stored at -20°C until use. The concentrations of IgG2a and anti-CII-specific IgG2a antibodies were measured using a mouse IgG2a ELISA quantification kit (Bethyl Lab Co., Montgomery, TX, USA). The absorbance at 405 nm was measured with an ELISA microplate reader (Molecular Devices).
Flow cytometry
For surface marker staining, single-cell suspensions were washed with FACS buffer and incubated with fluorochrome labeled-antibodies for 30 min at 4°C. For intracellular staining, single-cell suspensions were cultured with 25 ng/ml PMA (Sigma-Aldrich, p8139) and 250 ng/ml ionomycin (Sigma-Aldrich, I0634) with the addition of GolgiStop (BD Biosciences, #554715) for 4 h. After surface staining, cells were fixed and permeabilized with Cytofix/Cytoperm according to the manufacturer’s instructions (BD Biosciences, #554715). For intracellular Foxp3 staining, a Foxp3/Transcription Factor Staining Buffer kit was used (Invitrogen, #00-5523-00) after surface staining. After washing with Perm/Wash buffer, antibodies for intracellular staining were added for 30 min at 4°C. To identify murine Th17 cells, the cells were intracellularly stained with anti-IL-17-FITC Ab, followed by anti-CD4–PerCPCy5.5 Ab. To identify human Th17 cells, the cells were intracellularly stained with anti-IL-17-PE Ab, followed by anti-CD4-PE/Cyanine7 Ab. To identify human Treg cells, the cells were intracellularly stained with anti-Foxp3-FITC Ab, followed by anti-CD4-PerCP and anti-CD25-APC Ab. Stained cells were analyzed on a FACSCalibur (BD Biosciences) or CytoFLEX (Beckman Coulter). Events were collected and analyzed with FlowJo software (Tree Star).
Western blot analysis
Cells were lysed in Halt protein lysis buffer containing Halt phosphatase inhibitor (Thermo Scientific Pierce, Waltham, MA, USA), and protein concentrations were determined using the Bradford protein assay (Bio-Rad, Hercules, CA, USA). Proteins were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred onto Hybond ECL membranes (GE Healthcare, Waukesha, WI, USA), and incubated with antibodies against p-STAT3 (Tyr705, Ser727), STAT3, and β-actin or GAPDH for 10 min using the SNAP i.d. Protein Detection System (Millipore, Billerica, MA, USA). After washing, HRP-conjugated secondary Ab was added and incubated for 10 min. Ab binding was detected using an enhanced chemiluminescence detection kit (Pierce, Rockford, IL, USA) and Hyperfilm (Agfa, Mortsel, Belgium). Images were scanned on an Epson Perfection V700 photo scanner and quantification of intensity was performed using an ImageJ software. The levels of interest protein were normalized relative to that of β-actin or GAPDH as a loading control, and relative level of interest protein at nil condition was considered as 1.
Statistical analysis
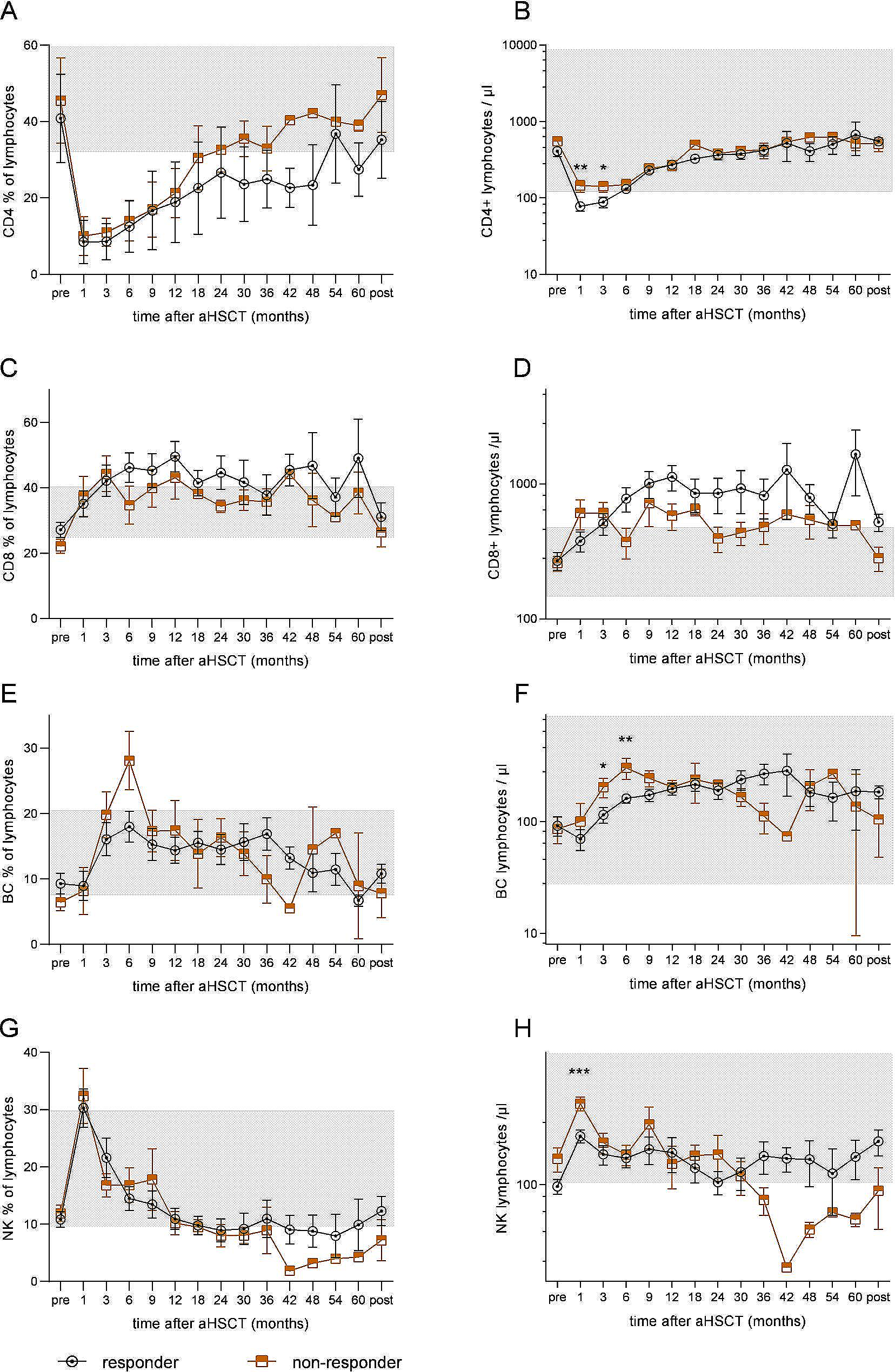
Statistical analyses were performed using GraphPad Prism, version 8 for Windows (GraphPad Software). P values were calculated using the two-tailed paired t-test and two-way analysis of variance (grouped). P < 0.05 was considered statistically significant. P values are presented within each figure or figure legend.

留言 (0)