
記住我

A 17-month-old male with trisomy 21, celiac disease, and history of complete atrioventricular septal defect status-post repair presented with persistent fevers, rash, hepatosplenomegaly, and serositis and arthritis on imaging. Labs revealed anemia (9.0 g/dL), thrombocytopenia (30,000), and elevated inflammatory markers, including ESR (81 mm/h), CRP (13.3 mg/dL), and ferritin (674 ng/ml). Initial investigation included a broad infectious workup, notably negative for CMV, EBV, adenovirus, parvovirus, hepatitis B, hepatitis C, and HIV. Immunological workup was largely unremarkable, with normal C3, C4, and IgG levels, along with normal NK cell function. IgA and IgM were mildly elevated at 225 mg/dL and 204 mg/dL, respectively. Initial cytokine testing included IL-6 only, which was elevated at 105 pg/mL. Hematological and oncological workup included bone marrow and inguinal lymph node biopsies which displayed no evidence of malignancy or HLH. Ultimately, a presumptive diagnosis of systemic juvenile idiopathic arthritis (sJIA) was made.
He therefore was promptly initiated on naproxen and a prednisolone bridge while beginning interleukin-1 (IL-1) inhibition via twice daily 2 mg/kg anakinra. He had several sJIA flares over the next several months manifested by recurrent fevers, rash, elevated inflammatory markers, and worsening arthralgias. Anakinra was discontinued and replaced with alternative IL-1 inhibition via monthly canakinumab. He also received maintenance prednisolone.
Despite the above therapies and dose adjustments, he continued to have frequent admissions for fevers and thrombocytopenia following infections. Finally, after an exhaustive infectious, malignant, and immune workup, the possibility of relapsing, refractory HLH/MAS was proposed. The diagnosis of HLH/MAS was ultimately made during an admission around 34 months of age and supported by persistent fever, hyperferritinemia (49,073 ng/mL), thrombocytopenia (59,000), anemia (8.4 g/dL), hypofibrinogenemia (50 mg/dL), elevated sIL-2r (13,079 U/ml), a highly specific marker for HLH [20], and hemophagocytosis visualized on repeat bone marrow biopsy. Supporting clinical criteria included transaminitis, hyponatremia, and associated coagulopathy. He was treated aggressively with high-dose methylprednisolone at 15 mg/kg twice daily for six total doses along with discontinuation of canakinumab and re-initiation of daily anakinra. The patient was discharged home on anakinra at 8 mg/kg daily and dexamethasone taper starting at 10 mg/m2/day over eight weeks per HLH-2004 protocol. Genetic testing for hereditary HLH, which included the four most common genes associated with familial HLH, PRF1, STX11, STXBP2, and UNC13D, along with several genes associated with X-linked lymphoproliferative disease and Griscelli syndrome, were all negative.
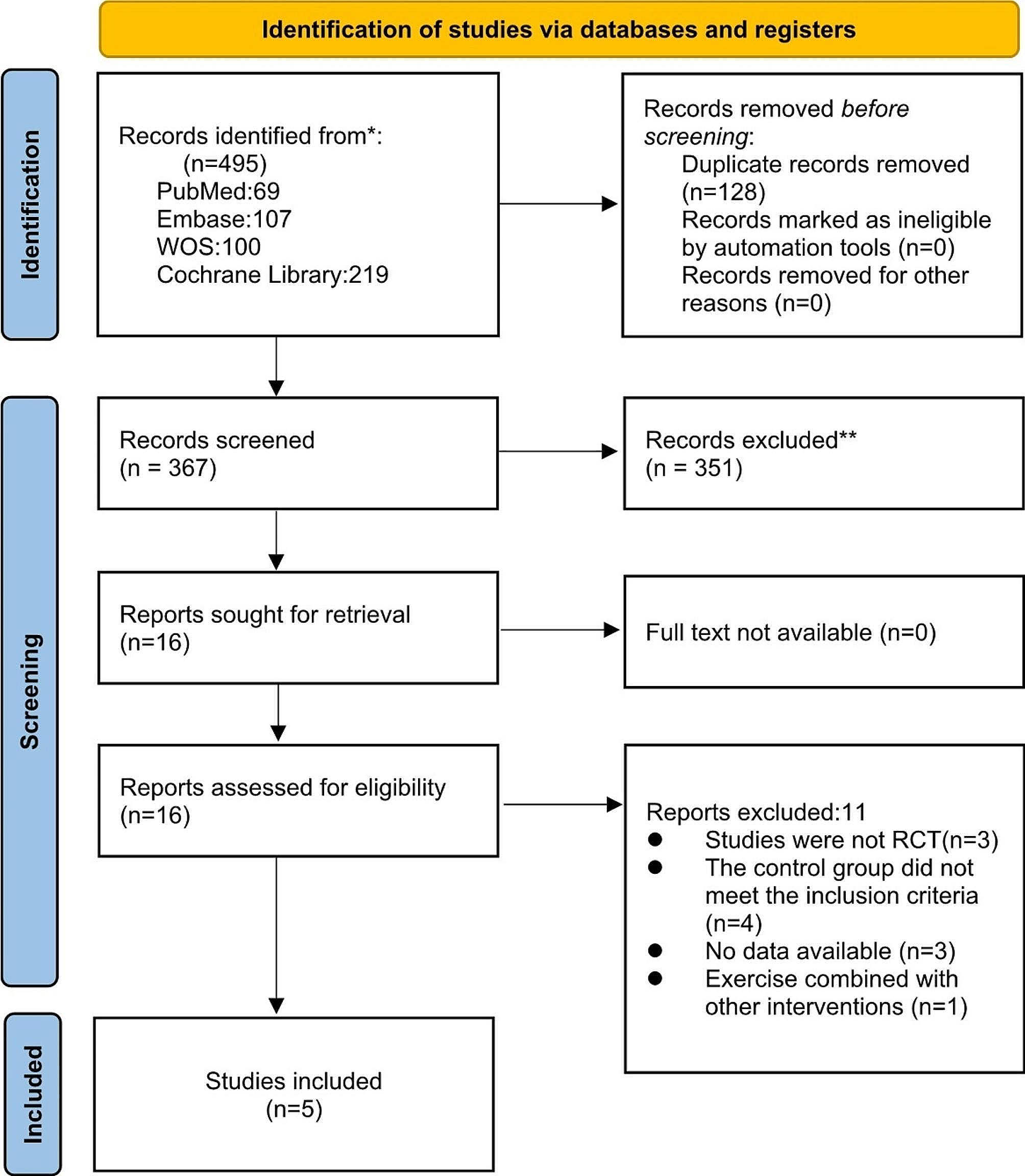
After six months of treatment, he again suffered a relapsing course. Given ongoing episodes of macrophage activation despite high doses of IL-1 inhibition, extensive multi-disciplinary discussions led to approved use of emapalumab, given its known efficacy in primary HLH, in addition to ongoing anakinra and dexamethasone. Emapalumab was administered twice weekly for 10 weeks, starting with 1 mg/kg and titrated up to maximum of 3 mg/kg. Several inflammatory markers, including ferritin, sIL-2r, and chemokine ligand 9 (CXCL-9), a chemokine induced by IFNγ, were frequently monitored (see Fig. 1). While on emapalumab, the patient had both clinical and laboratory improvement. The patient was weaned off emapalumab and transitioned to twice daily baricitinib at 2 mg per dose (0.26 mg/kg/day). The decision to transition to baricitinib was largely due to cost efficacy and easier administration in that the patient could avoid long-term need for infusions. Baricitinib has been efficacious in treatment of interferonopathies given its suppression on IFNγ, so was trialed as maintenance in this patient, which ended up also being efficacious.
Fig. 1
Various labs, including ferritin, soluble interleukin 2 receptor (sIL2-r), and chemokine ligand 9 (CXCL-9) values were trended over four years. The maximum values for a chosen time frame are included
He did require re-hospitalization at 5 years of age for acute respiratory failure secondary to COVID-19 pneumonia. There was associated hyper-inflammation but the patient did not develop HLH/MAS at that time. He was maintained on baricitinib and anakinra throughout the illness. He did require a short course of corticosteroids for respiratory indications during hospitalization, which were able to be weaned off without relapse of hyper-inflammation or HLH/MAS. To date he continues to do well on maintenance anakinra and baricitinib without additional disease flare for over two years.
Case 2A 15-month-old female with trisomy 21, recurrent otitis media, persistent fevers and rash, and recurrent thrombocytopenia was admitted with fever, emesis, and acute liver failure. Initial infectious workup was negative for acute CMV, EBV, adenovirus, parvovirus, HSV, and HIV. Bone marrow biopsy was negative for malignancy and without evidence of hemophagocytosis. After an extensive infectious, malignant, and immune workup, she was diagnosed with a form of steroid-dependent HLH. Diagnosis of HLH was made based on fevers, hyperferritinemia (6,838 ng/ml), pancytopenia (WBC 1,900 though ANC > 1000, Hgb 6.2 g/dL, platelets 44,000), hypofibrinogenemia (96 mg/dl), elevated sIL-2r (12,390 U/ml), elevated sCD163 (8878 ng/mL) and decreased NK cell count and function with poor cytotoxicity, as evidenced by low CD107a, which can be used as a marker for NK cell activity. Additionally, she had decreased percentage of NK cells expressing perforin. Supporting clinical criteria for the diagnosis for HLH included transaminitis, vomiting, and weight loss. Her initial presentation was complicated by Streptococcus pneumoniae bacteremia and DIC. The patient was initiated on anakinra, along with an eight-week dexamethasone taper and etoposide, though delayed initially due to liver injury, per HLH-2004 protocol. Genetic testing for hereditary HLH, which included 14 associated genes, was negative. The etiology of her HLH was initially suspected to be secondary to an infectious trigger or underlying primary immunodeficiency given her low NK cell count and poor function. Of note, on initial presentation, she had normal immunoglobulin levels and normal pneumococcal serologies, suggesting functional B cells.
By three years of age, she continued to have repeated flares of macrophage activation, mainly triggered by attempts at weaning steroids as well as frequent infections, including viral sources, MRSE bacteremia, Clostridium difficile colitis, abdominal abscess, and cellulitis. Given her frequent exacerbations with steroid-dependence and despite high doses of IL-1 inhibition with both anakinra and canakinumab trials, she was initiated on emapalumab at 1 mg/kg twice weekly. Emapalumab was chosen given its efficacy in primary HLH. It was proposed that her HLH was due to underlying immune dysregulation and hyper-interferon signaling associated with trisomy 21. A baseline CXCL-9 was elevated at 303 pg/ml, affirming increased IFNγ signaling. While on emapalumab, ferritin, sIL-2r, CXCL-9, and other inflammatory markers were frequently monitored (see Fig. 2). She had both clinical and laboratory improvement so twice daily baricitinib was initiated at 2 mg per dose (0.3 mg/kg/day) after about five weeks of emapalumab treatment. Continued stability allowed successful wean of emapalumab after ten total weeks of treatment. She has continued to do well without return of disease for three years on maintenance baricitinib, allowing successful withdrawal of corticosteroids for the first time since diagnosis, and an ongoing slow wean of anakinra.
Fig. 2
Ferritin, soluble interleukin-2 receptor (sIL2-r), and chemokine ligand 9 (CXCL-9) values were trended over four years. The maximum values for a chosen time frame are included on above graph
留言 (0)