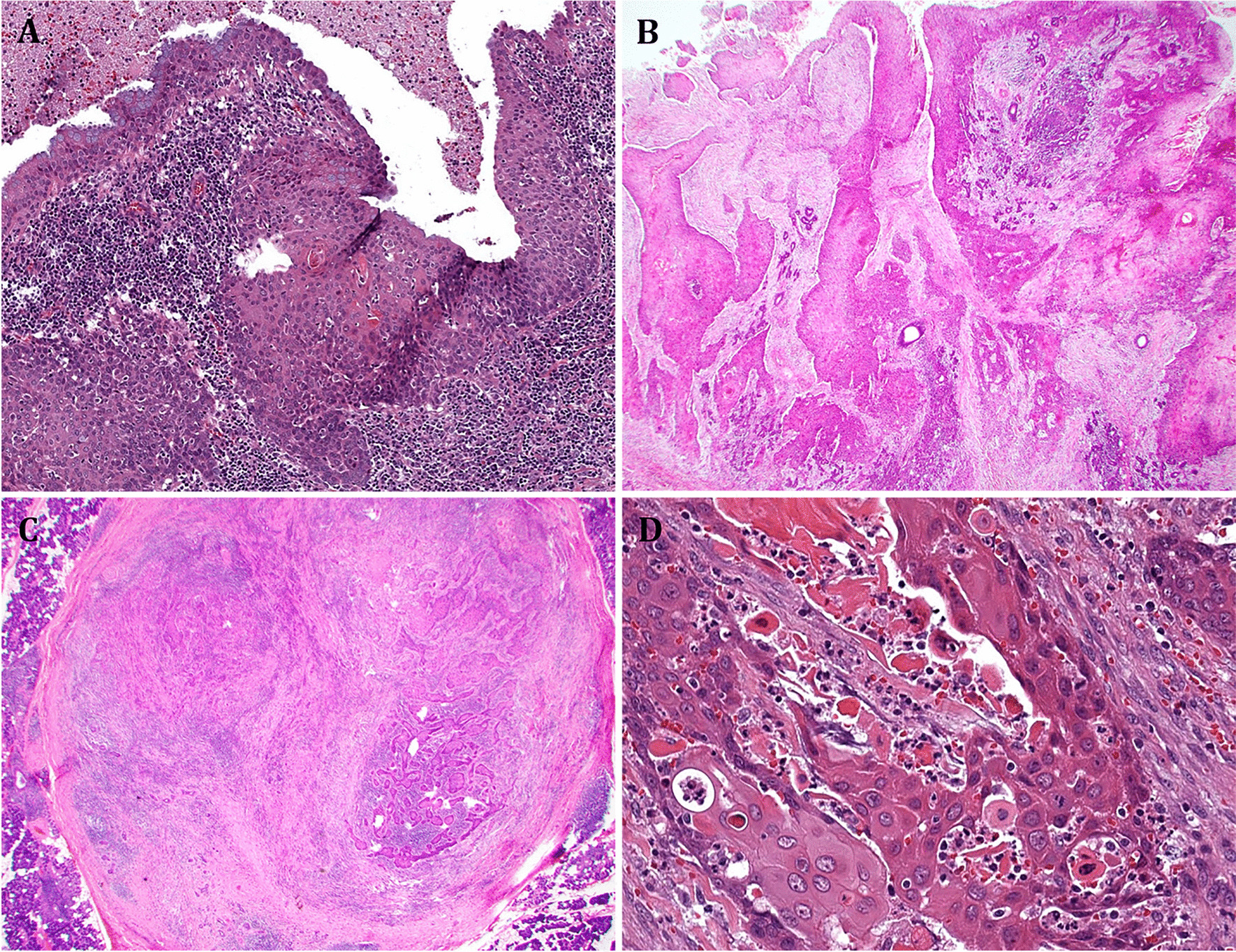
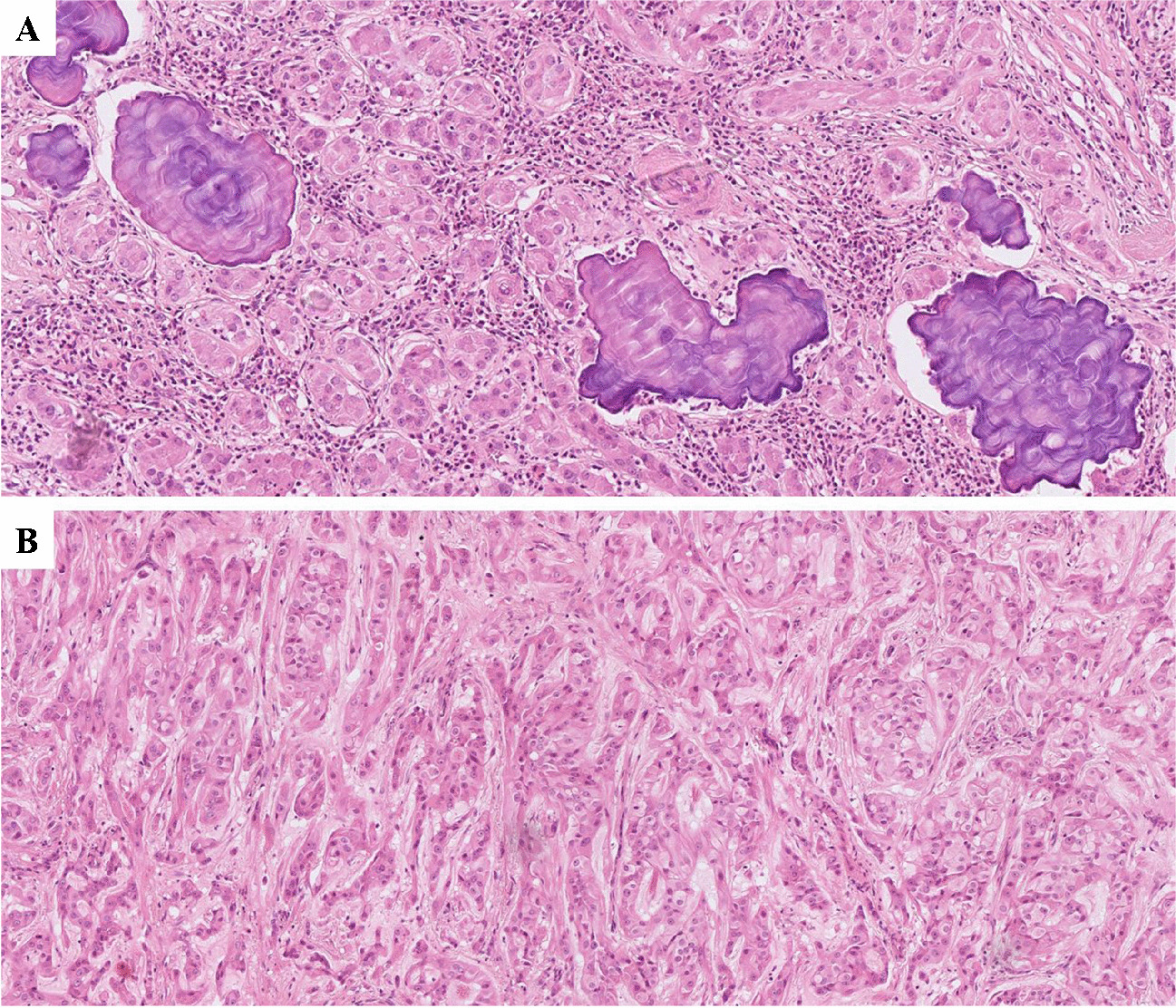
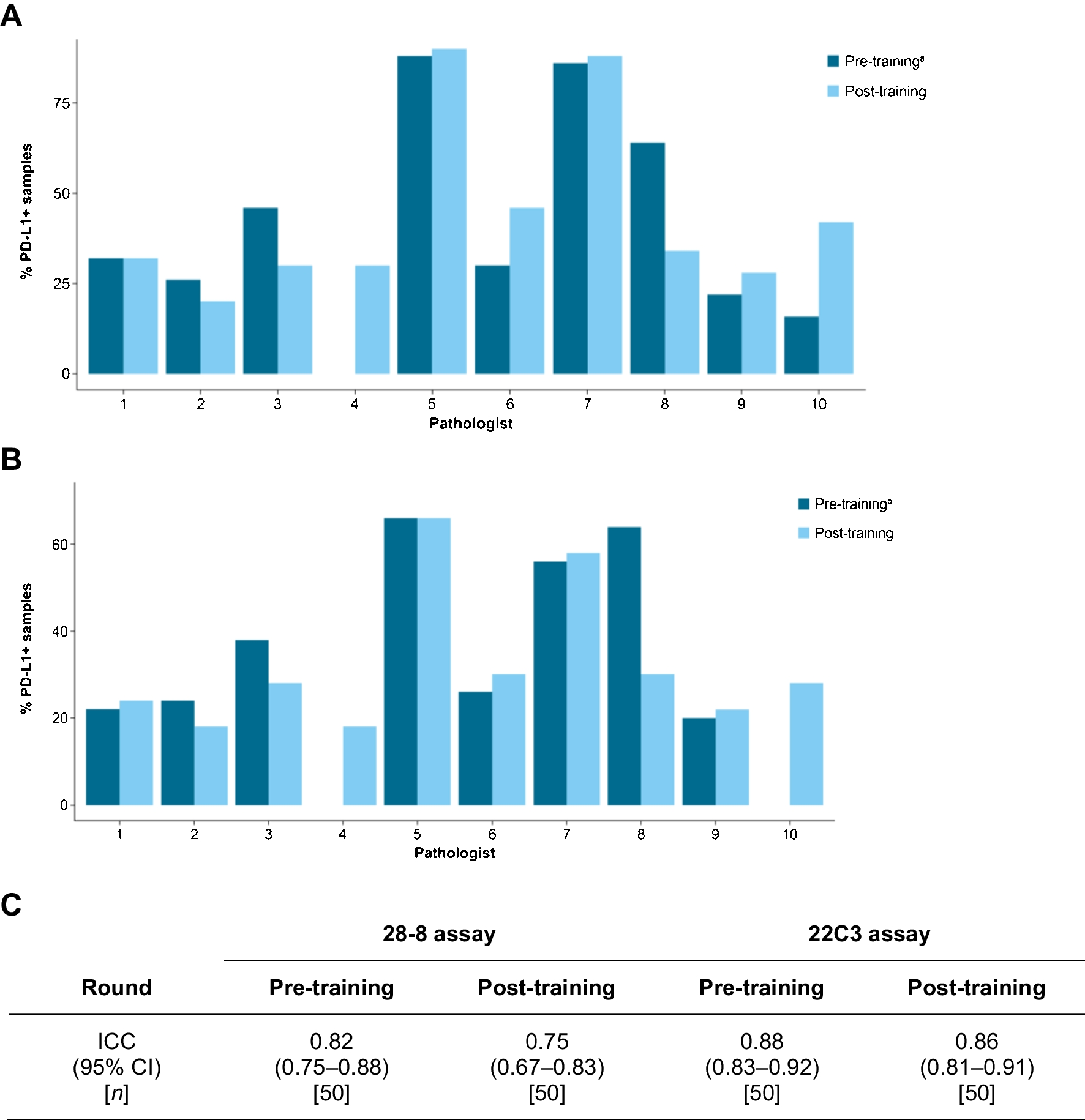
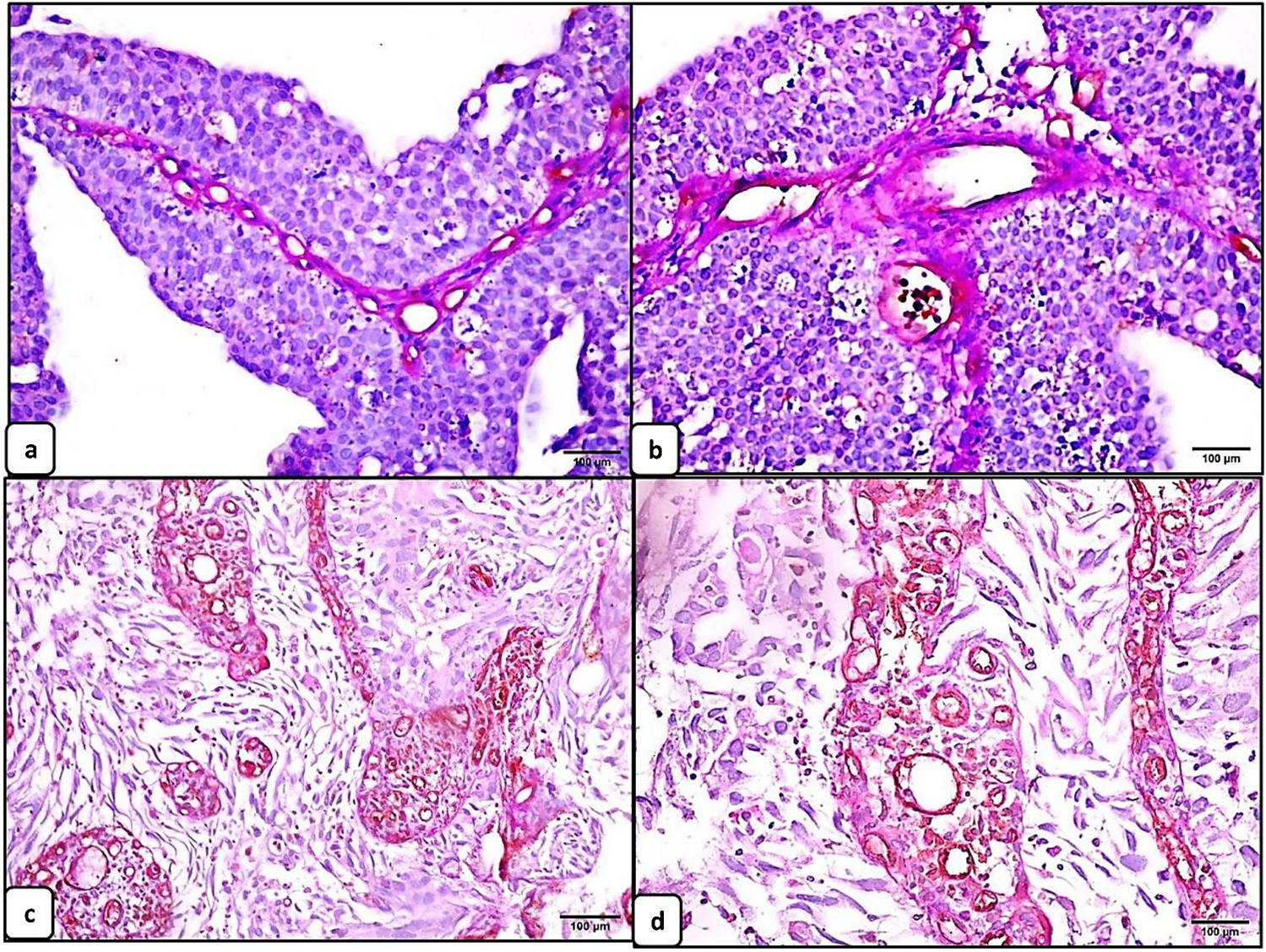
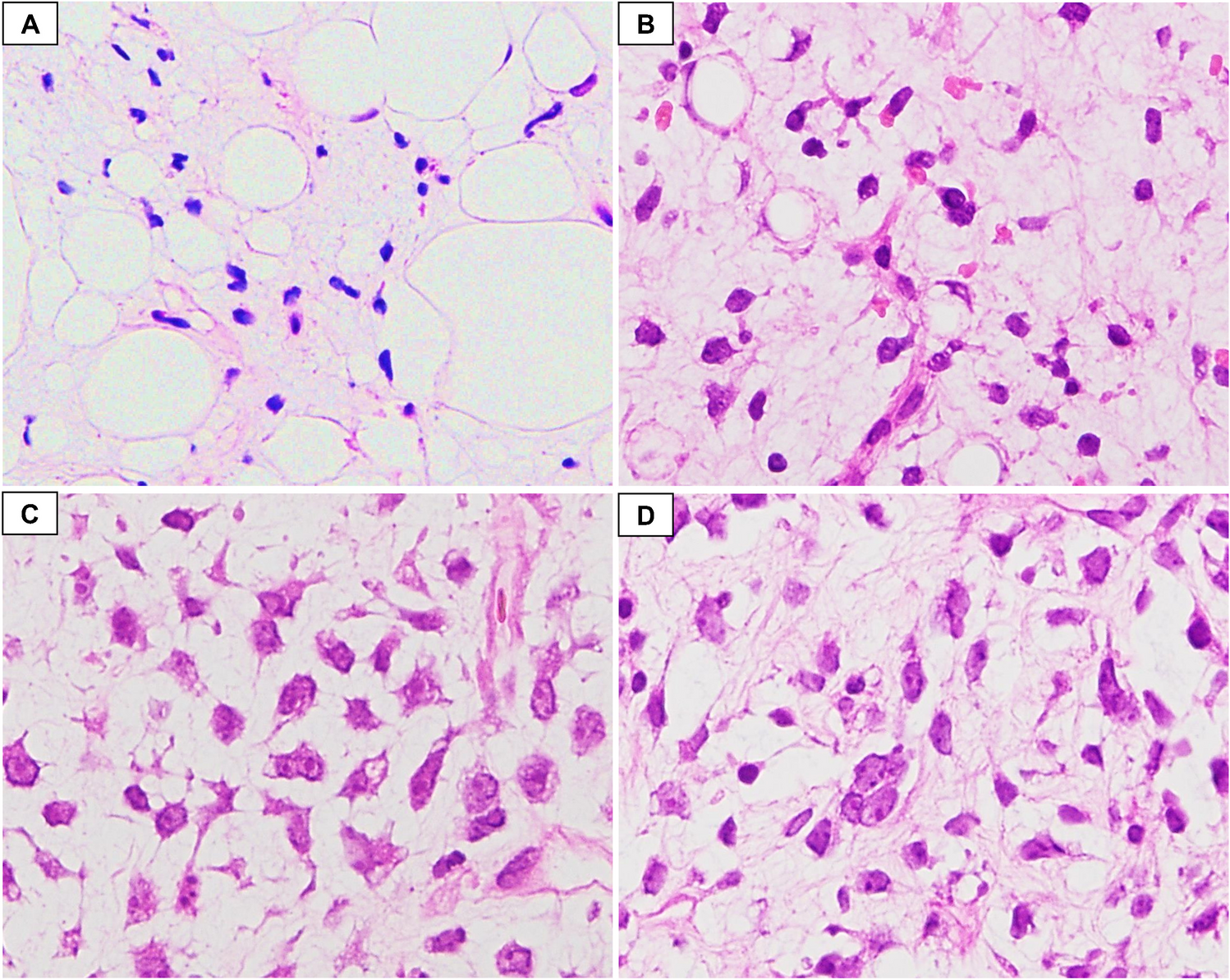
Granulosa cell tumour of the ovary is often described as an incidental finding in only one ovary after hysterectomy with bilateral adnexectomy in the setting of atypical endometrial hyperplasia with bleeding, rarely associated with endometrial carcinoma [2]. Morphologically, the tumour is characterized by (a) a multinodular growth pattern, often with (b) one bigger bulging nodule, and (c) a sharp yellow cut surface. Microscopical characteristics comprise (d) areas of extensive hyalinized sclerosis with (e) compressed strands of inconspicuous small spindle cells with angulated nuclei, and (f) intermingled nests of epithelioid thecoma-like cells with pale eosinophilic cytoplasms and small, round, or angulated nuclei with nuclear grooves [2] (Fig. 1, Table 1). Immunohistochemistry corresponds to typical sex cord-stromal tumour profile (positivity for SF1, calretinin, inhibin, vimentin, often FOXL2; often oestrogen and progesterone receptor expression; and negativity or weak patchy staining for keratin, negativity for EMA, PAX8, and PAX2) (Fig. 2). Most important and diagnostically relevant is the reticulin staining [2]. Significant reticulin loss highlights small nests of thecoma-like cells embedded in extensive sclerosis. In contrast to true thecoma cells, these thecoma-like nests do not present the typical argyrophilic single cell ensheathment classically observed in thecoma. A prominent fibre component in sclerotic areas may be seen, but reticulin staining is strongly reduced or lost in both in the granulosa cell and the thecoma-like foci, demonstrating the characteristic nodular tumour architecture. The reticulin staining is a simple, helpful, and a quick method to foreground features of a tumour in suspect-appearing sclerotic ovaries.
Adult granulosa cell tumour (AGCT) belongs to the group of pure sex cord-tumours (deriving from granulosa or Sertoli cells). Juvenile granulosa cell tumour (JGCT), arising in children and young adults, represents a clinico-pathologically distinctive entity that differs from AGCT in many aspects. In opposition, the current WHO classification lists the group of pure stromal tumours, originating from stromal cells (e.g. fibroma, thecoma), and the group of mixed sex cord-stromal tumours with aspects of both (e.g. Sertoli-Leydig cell tumour). However, as broadly described in the literature since the 1930s by Traut et al. [3], mixed forms between AGCT and fibrothecoma arise, with varying compounds of both entities in one neoplasm. The final tumour classification as ACGT, fibrothecoma or a mixed form, depends on the respective degree of different tumour cells. In 1968 and 1977, Norris et al. [4] and Stage et al. [5] described tumours with a percentage of granulosa cells between 10 and 50% as granulosa cell tumours or mixed granulosa theca cell tumours. Young et al. considered fibroma/thecoma with a granulosa cell component less than 10% as benign “fibrothecomas with minor sex cord elements” [6]. Tumours with more than 50% of granulosa cells were supposed to correspond to AGCT. In a more recent review (2014), Oliva and Young classified tumours with more than 10% granulosa cells on a fibrothecomatous stroma as granulosa cell tumours [7].
Generally, ACGT occurs predominantly in postmenopausal women (peak incidence 50–55 years) [8,9,10]. As the most common functionally active, oestrogenic ovarian tumour, it often presents with uterine bleeding caused by tumour-induced (atypical) endometrial hyperplasia, endometrial polyps [11,12,13], and rarely by endometrium carcinoma [1, 6, 14]. Only occasionally an androgenic or progestogenic effect is observed. There are only a few reported cases in the literature describing AGCT arising from extraovarian tissue, mainly from the broad ligament [15]. AGCT is a low malignant tumour with potential of (often late) recurrence and metastasis in 20–30% even in early stages [16, 17], sometimes decades after the initial diagnosis [11, 14, 18]. Documented 10-year overall survival varies. Older studies report survival rates from less than 60% to 90%. However, a recent study from 2016 by McConechy et al. found that 10-year overall survival was identical to the general population, with a median time of recurrence of 7.2 years [19]. A study from 2015 by Wilson et al. reported a relapse of one-third of stage I tumours with a medium relapse time of 12 years [20]. Several long-term studies from the late 1970s and early 1980s also demonstrated late progression [9, 10, 21, 22], with the latest documented relapse after 37 years [19]. It is presumed that lower survival rates might be due to the inclusion of misdiagnosis (in one study, in more than 50% of the cases [12]). Histopathologic errors include metastatic lobular breast carcinoma, malignant melanoma, epithelioid mesothelioma, as well as small cell carcinoma of the hypercalcaemic type (OSCCHT) and clear cell carcinoma [7, 13], which are considered as the most important, albeit rare, differential diagnosis of AGCT [11]. Juvenile granulosa cell tumour must be mentioned as differential diagnosis with a more favourable curse. Luteinized subtype of AGCT can be mistaken as a steroid cell tumour. Again, reticulin staining is helpful, highlighting single cell ensheathment of the latter [13].
In 2008, Shah et al. demonstrated FOXL2 c.402C \(\to\) G (p.C134W) mutations in 95–97% of pure adult granulosa cell tumours [23], considered as a good molecular discriminator in diagnostically uncertain cases [24]. FOXL2 encodes Forkhead Box L2 (FOXL2), suggested to act as a tumour suppressor in granulosa cells mediating apoptosis. Pathogenic FOXL2 mutation is discussed to cause imbalances in the transforming growth factor β (TGFβ)-signal pathway via impaired interaction with SMAD transcription factors [25, 26]. However, especially in mixed forms with aspects of fibrothecoma, FOXL2 mutation can be negative [2]. Nolan et al. found FOXL2 mutations in six of twelve mixed granulosa theca cell tumours with a granulosa tumour cell component greater than 30% [27]. In five cases, FOXL2 mutations were detected via microdissection in both the granulosa cell and the thecoma cell component. McClugagge et al. reported FOXL2 mutations in so called “cellular mitotically active fibromas with epithelioid nodules” but not in cellular fibromas [24], and Shah et al. described a FOXL2 mutation in a “thecoma with minor granulosa cell component” [23]. These findings evoke the question if these cases, in fact, corresponded to mixed forms of granulosa cell tumour with a significant and predominant fibrothecomatous component, further supporting the consideration that on a molecular basis, mixed tumours with a minor percentage of granulosa cells might be more consistent with granulosa cell tumour.
Prognostic factors are difficult to determine. To date, only the International Federation of Gynaecology and Obstetrics (FIGO) stage appears to be the single reliable and reproducible factor to estimate risk of recurrence or metastatic dissemination [18, 28,29,30]. Ninety percent of AGCT present at FIGO stage I with good prognosis (90% 5-year overall survival); stage IV is a rarity [12, 31]. Some studies discuss tumour size as a possible risk factor [14, 17]. Tumours up to 5 cm in diameter were shown to have far better overall survival than bigger tumours from 6 to 15 cm, 10 to 15 cm [14], or 13.5 cm [17]. Bigger tumours tended to be less solid but more cystic and haemorrhagic, with an elevated risk of rupture and haematoperitoneum, hence an increased danger of dissemination and recurrence. In contrast to that, other studies did not find a correlation between tumour size and risk of recurrence, but between tumour stage and elevated mitotic rate [12, 31]. On a molecular basis, one extended Finnish study demonstrated a correlation between long-term, disease-free survival and high-level, zinc-finger transcription factor GATA4 and human epidermal growth receptor HER2 expression [32], and FOXL2 [33], as well as nuclear atypia [34]. This was already described before in 2005 by Anttonen et al. [35, 36] and by Leibl et al. [37]. GATA4 interacts with SMAD3, a member of TGFβ signal cascade. The impact of “nuclear atypia” appears questionable, since other studies describe AGCT with bizarre, “symplastic” nuclei as a degenerative phenomenon without impact on prognosis [6, 13].
Low tumour stage IA requires mere surgical resection with hysterectomy and bilateral salpingo-oophorectomy. If young patients wish to have children, unilateral salpingo-oophorectomy is sometimes possible. A standard therapy for higher tumour stages (IC–IV) or intraoperative rupture does not exist. Defined risk factors for disease relapse are unknown, apart from tumour size and spontaneous or intraoperative rupture. Age at diagnosis does not appear to play a role. Pelvic and retroperitoneal lymphonodectomy and omentectomy may appear obligative in certain cases, adapted to treatment of epithelial ovarian cancer. However, lymph node metastasis and haematogenous spreading is a very rare finding; AGCT rather seeds via peritoneal dissemination [37]. A recent study from 2019 did not find a benefit in lymphonodectomy on overall survival, but a negative effect in surgical outcome, independent from tumour stage [31]. Most tumours recur in the pelvic or abdominal region, seldomly in the liver and bone [20]. Chemotherapy for recurrent and disseminating tumours [38] is platinum or taxane based, possible combined regiments comprise bleomycin, vinblastine or alternatively etoposide, and cisplatin (BVP/BEP) [20, 39]. Furthermore, antihormonal therapies with luteinizing hormone-releasing hormone (LHRH)-antagonists, tamoxifen, aromatase inhibitors, gonadotropin-releasing hormone (GnRH)-analogist leuprorelin, or the progesterone derivate megestrol show good responses in recurring oestrogen secreting tumours [20, 40]. Other modern targeted therapies are considered, especially trials with bevacizumab as an inhibitor of vascular endothelial growth factor [41,42,43,44], and the tyrosine kinase inhibitor, imatinib [45]. EGFR targeting may be considered in a subgroup of EGFR positive tumours [37, 46]. Radiation therapy can be an option if surgery is not possible.




留言 (0)