
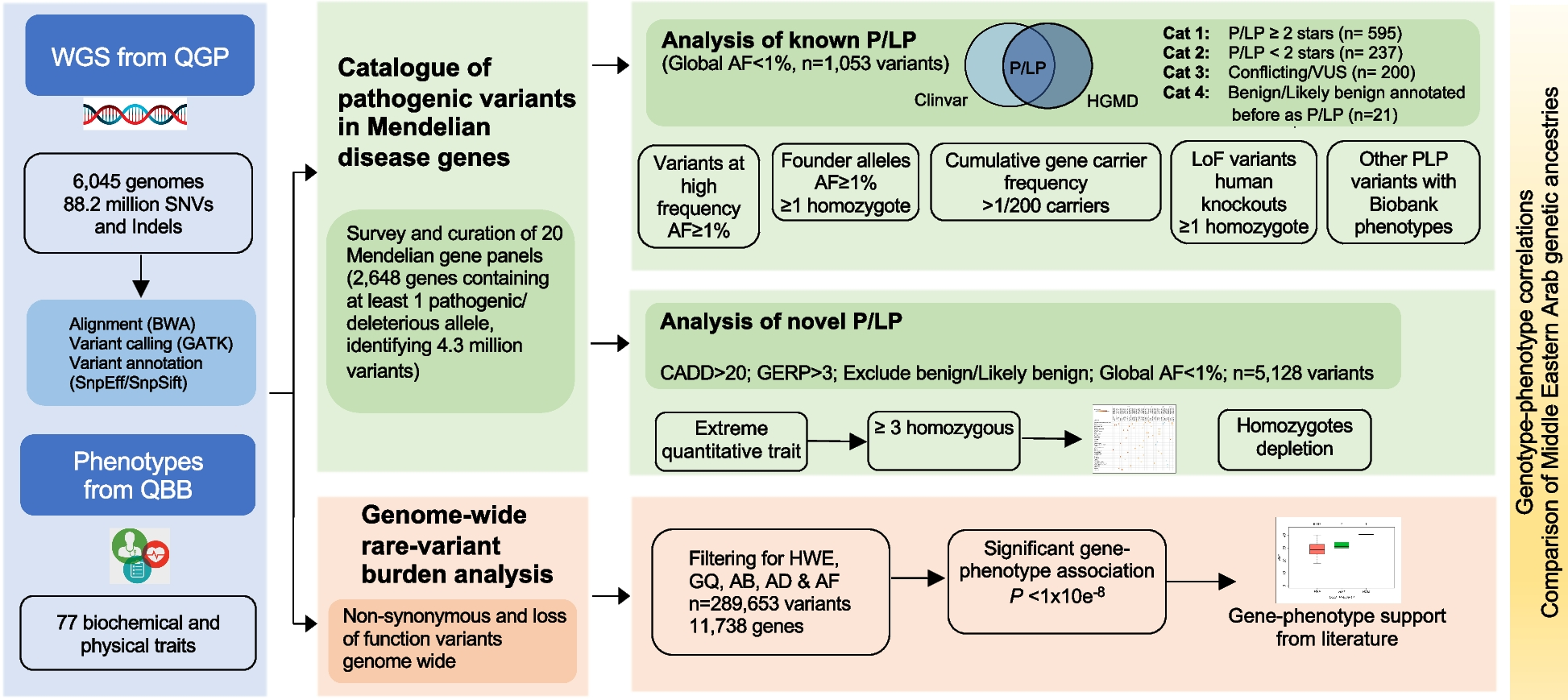
記住我
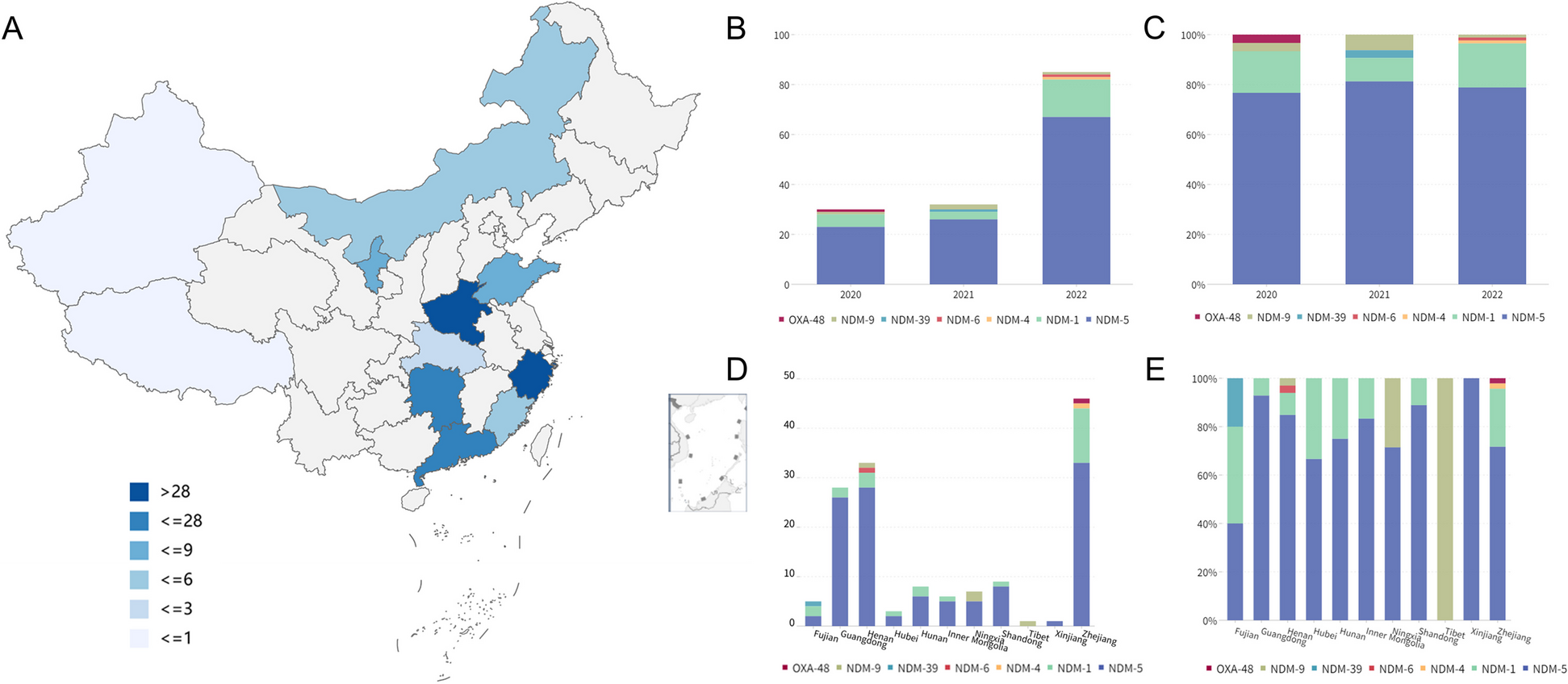
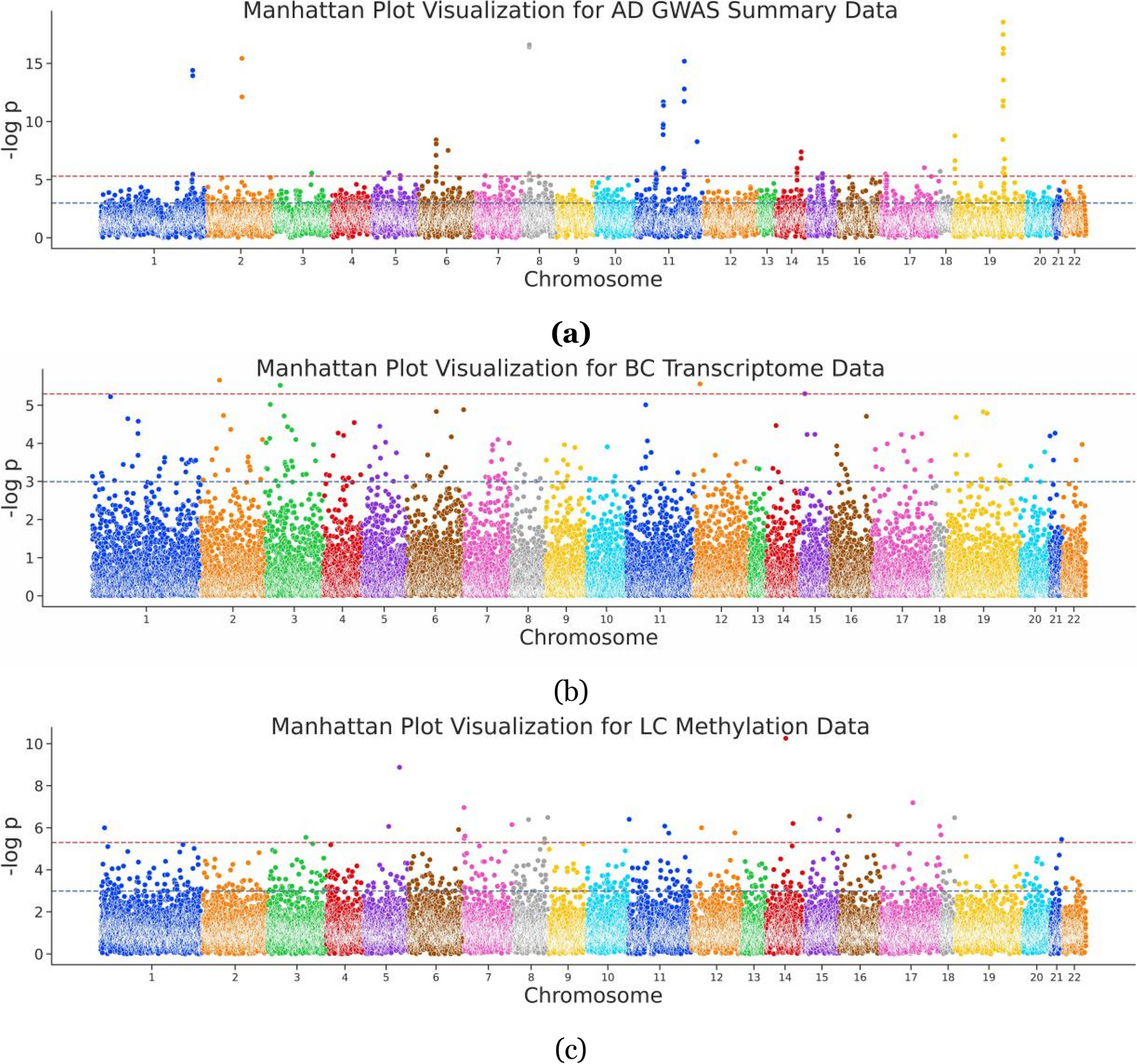
Examining first a cohort of 842 Parkinson’s disease patients (Oxford Discovery cohort [4, 6]) which had been genotyped and phenotypically characterised with 40 clinical assessments (Additional file 2: Table S1), we applied the PHENIX MPMM method to identify underlying latent continuous phenotypic axes that could account for the observed clinical variation. Each phenotypic axis reflected a number of co-varying observed clinical assessments. Three phenotypic axes explained more than 75% of the clinical variation, specifically Axes 1, 2 and 3 explained 39.6%, 28.7% and 6.8% of the variation respectively (Fig. 2 and Additional file 1: Fig S5). To examine whether similar phenotypic axes are obtained in different deeply phenotyped Parkinson’s disease cohorts, we derived phenotypic axes within an independent cohort of 1807 Parkinson’s disease individuals from the Tracking UK cohort [2] that had made similar clinical observations to the Oxford Discovery cohort [4, 6]. We found significant Pearson’s correlation coefficients between each cohort’s first three phenotypic axes: Axis 1 r = 0.92 (p = 3 × 10−13), Axis 2 r = 0.89 (p = 4 × 10−11), Axis 3 r = 0.72 (p = 5 × 10−6) (Fig. 2). Nevertheless, a major concern was that the identification of the same phenotypic axes might, at least in part, be due to the very similar structure of the clinical phenotyping between the two UK cohorts. To address this, we examined the independent US-based PPMI cohort [10] consisting of 439 sporadic Parkinson’s disease individuals that had been clinically phenotyped following a substantially different protocol to the UK cohorts. After deriving phenotypic axes in the PPMI cohort [10], we found significant similarities between the first three phenotypic axes derived for both Oxford Discovery [4, 6] and PPMI [10] cohorts: the coefficients of determination (R^2) between three first axes across different categories of clinical phenotypes from each cohort were: Axis1: 0.665 (p = 0.048), Axis 2: 0.914 (p = 0.003) and Axis 3: 0.754 (p = 0.025) (Fig. 3 and Additional file 1: Fig S6). By deriving phenotypic axis in three cohorts by using only UPDRS I, II, III and MOCA, four clinical measures systematically recorded in each cohort, we found significant similarities between the two first phenotypic axes derived in three cohorts: correlation between phenotypic axis vs clinical measure between Oxford Discovery cohorts (x-axis) vs others cohorts r = 0.92 95% [0.81–0.97]. These consistent similarities in the axes of phenotypic variation independently derived for each of three different Parkinson’s disease cohorts demonstrates the universality of these axes of phenotypic variation amongst Parkinson’s patients. Finally, by comparing PHENIX with other methods of dimensionality reduction for the UK/US cohort comparisons, specifically principal component analyses (PCA), multidimensional scaling (MDS) and independent component analysis (ICA), only the phenotypic dimensions discovered by the genetically-guided MPMM model, PHENIX, were significantly correlated between both cohorts. Hence, no other method was able to identify similar axes of phenotypic variation across UK and US Parkinson’s disease cohorts (Fig. 3).
Fig. 2
Similar phenotypic axes are obtained in two deeply phenotyped Parkinson’s disease cohorts. Results were consistent in two independent cohorts (842 Oxford Discovery and 1807 Tracking UK patients). Examination of these two separate Parkinson’s disease cohorts, using an independent derivation of the phenotypic axes in each, showed significant correlations between each cohort’s first three axes. Correlations between the axes from each cohort are Axis 1 r = 0.92 (p = 3 × 10–13), Axis 2 r = 0.89 (p = 4 × 10−11), and Axis 3 r = 0.72 (p = 5 × 10−6). The correlation coefficient (x-axis) between each axis derived in each cohort (light: Oxford Discovery vs dark: Tracking UK) and each clinical observation (y-axis) is shown. We represented six major categories of Parkinson’s disease symptoms by the colour of the bar plots. These categories include anxiety and depression, the autonomic system, cognitive functions, the motor system, the olfactory system and sleep disorders. The Unified Parkinson’s Disease Rating Scale (UPDRS) is a comprehensive 50-question assessment of both motor and non-motor symptoms associated with Parkinson’s. It includes four parts: (I) non-motor experiences of daily living (II) motor experiences of daily living (III) motor examination (IV) motor complications
Fig. 3
Other methods fail to align between different but deeply phenotyped UK and US Parkinson’s disease cohorts. We compared the ability of different dimensionality reduction methods (independent component analysis (ICA), multidimensional scaling (MDS), principal component analysis (PCA) and phenotypic axis based on the PHENIX multiple phenotype mixed model) to phenotypically align two deeply phenotyped Parkinson’s disease cohorts, specifically the Oxford Discovery (842 individuals) and PPMI (439 sporadic Parkinson’s disease) cohorts. The x-axis and y-axis represent the correlation coefficient between each continuous variable with clinical observation associated with a specific symptom category in Oxford Discovery and PPMI cohort, respectively. Each column panel and colour of points (“Axis”) represents the dimension level of each underlying dimension. All points on the diagonal would represent a perfect phenotypic alignment of both cohorts. We examined the relationship between correlation derived from both cohorts by performing a linear regression: R^2 and p correspond to the coefficient of determination and the p-value respectively. Only the dimensions discovered by the MPMM model, PHENIX, show a significant relationship between both cohorts: MPMM phenotypic axes (R2 = 0.86, p = 2 × 10−8), MDS (R2 = 0.11, p = 0.18), ICA (R2 = 0.17, p = 0.16) and PCA (R.2 = 0.31, p = 0.06)
Each phenotypic axis represents a distinct set of clinical featuresTo interpret the clinical relevance of each phenotypic axis, we examined the correlation between individual clinical features and the phenotypic axes (Table 2 and Additional file 1: Fig S2 and Additional file 1: Fig S7). We observed that each phenotypic axis corresponded to a subset of clinical features, differing in both extents and directions of severity. Axis 1 represented worsening non-tremor motor phenotypes, anxiety and depression accompanied by a decline of the cognitive function (Table 2). Worsening anxiety and depression were also features of Axis 2, in addition to increasing the severity of autonomic symptoms and increasing motor dysfunction. Axis 3 was associated with general motor symptom severity including rigidity, bradykinesia and tremor of the whole body independently of non-motor features. The contribution of different phenotypes to these axes was therefore highly variable. Specific aspects of motor dysfunction were important factors in defining the majority of axes. Anxiety and depression were also relatively important features, but only for axes explaining the largest amounts of variation. Conversely, cognitive impairment was associated only with Axis 1. However, this observation must be weighted by the fact that cognitive impairment/dementia is reported at a later disease stage and thus features less in recently diagnosed cases.
Table 2 Correlation between each axis and each clinical phenotypic measureAlthough each phenotypic axis is associated with a distinct set of clinical features, they are not independent but instead strongly correlated (Additional file 1: Fig S8). We find no significant relation between the phenotypic axes and principal components of genetic ancestry (Methods) suggesting that the phenotypic axes are not biased by the population structure (Fig S9, Additional file 2 :Table S5). However, as previously reported, gender influences clinical symptoms [4] and we also observe a significant association between gender and Axis 2 (Table S5, p = 4.5 × 10−5).
To assess to what extent the phenotypic axes might be affected by the number of clinical observations, within the Oxford Discovery cohort [4, 6] we compared the phenotypic axes built on all clinical features with phenotypic axes generated with incomplete sets of randomly-selected clinical features. We observed a strong correlation (r > 0.8) between each of the two first phenotypic axes built with as few as 50% of the clinical variables and their respective original phenotypic axes, suggesting that these two axes are extremely robust in terms of the numbers of clinical variables considered (Additional file 1: Fig S9). Finally, the agreement of these phenotype axes with previously observed correlations provides further support for underlying biological themes, but their reinterpretation as robust continuous traits likely provides a more realistic approximation of how the underlying biology contributes, as opposed to a clustering-based cut-off for a phenotype. Specifically, the unimodal distribution of patients along these phenotypic axes (Additional file 1: Fig S10 and S11) suggests here that the development of continuous measures is more appropriate than clustering according to an arbitrary threshold.
The integration of genetic relationships improves the capture of the clinical symptomsThe PHENIX MPMM approach employed here to derive phenotypic axes exploits the genetic relatedness between individuals derived from genotypic similarity to further decompose random effects into kinship effects between individuals. In its original application to imputing missing phenotypes, PHENIX outperforms other imputation approaches when the heritability (h2) of a phenotype increased [9]. Similarly, when randomly removing and re-imputing 10% of observed data, the quality of the imputation of Parkinson’s disease clinical assessments was in general better when considering the genetic relatedness between individuals as compared to excluding this information (Additional file 1: Fig S12), suggesting that phenotypic axes better capture Parkinson’s disease heterogeneity when including genetic information. Moreover, we found a higher agreement between the phenotypic axes derived by integrating the genetic relationship between patients of different cohorts than when the phenotypic axes were derived ignoring the genetic relationships (Additional file 1: Fig S13). Specifically, the coefficient of determination reflecting the agreement between the axes derived from Oxford Discovery [4, 6] and those derived from the PPMI [10] cohorts were from Axis 1 to 3, respectively: 0.665 (p = 0.048), 0.914 (p = 0.003) and 0.754 (p = 0.025) when including the genetic similarity between patients as compared to 0.604 (p = 0.069), 0.908 (p = 0.003) and 0.001 (p = 0.991) without. Together, these findings demonstrate that including genetic relationships between patients enhances the resulting phenotypic axes’ ability to reproducibly capture Parkinson’s disease clinical variation.
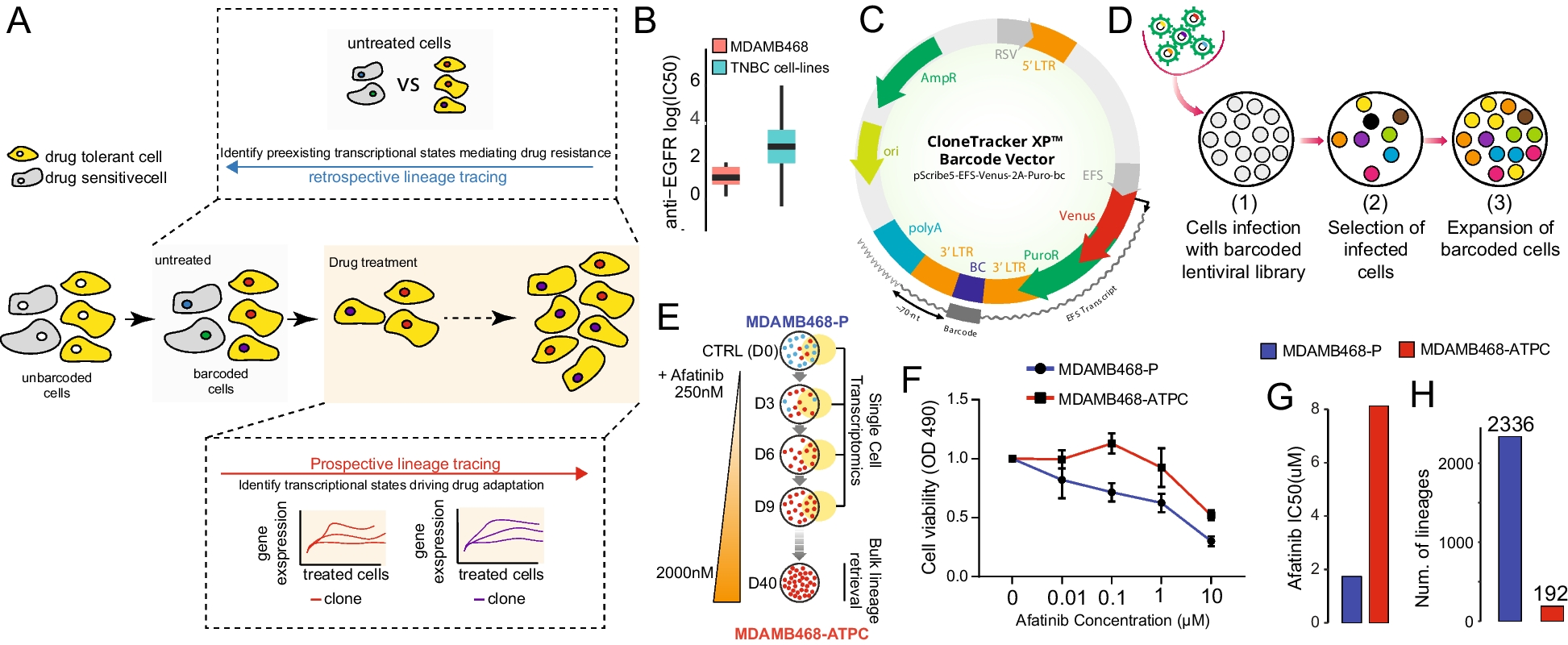
A high Alzheimer’s genetic score increases the risk of developing a more severe Parkinson’s formTo better understand the genetic risk factors influencing the phenotypic axis, we replaced the pairwise patient overall genotypic similarity matrix in the MPMM with a similarity matrix based only on regions of the genome associated with a specific complex human trait/disease. For example, replacing the overall genetic similarity with how similar people are in their genetic risk for diabetes or depression. We then rederived the phenotypic axes using the new metric of genetic similarity and compared the proportion of phenotypic variation explained by the new phenotypic axes, derived from different disease risks, to the original phenotypic axes that were derived using the entire genotype (Methods). Unexpectedly, the phenotypic axes derived using Parkinson’s disease genetic risk performed no better than the original phenotypic axes, while axes derived using the genetic risk for Alzheimer’s disease or the risk for inflammatory bowel disease, ulcerative colitis significantly outperformed, i.e. captured more patient phenotypic variation than, the original principal phenotypic Axis 1 (Fig. 4A and Additional file 1: Fig S14-15). Although UC and inflammatory bowel disease share a common genetic aetiology [23], we find no evidence that the same risk variants influence Alzheimer’s disease, suggesting that two distinct molecular aetiologies underlie phenotypic Axis 1. Specifically, we see no significant reduction in the variance explained by the axis calculated using Alzheimer’s disease genetics variants conditioned on ulcerative colitis or inflammatory bowel disease genetics variants (Additional file 1: Fig S16). The APOE locus is one of the major risk loci in Alzheimer’s disease, but we found no evidence that Parkinson’s disease individuals carrying one of two APOE ε4 alleles have a significantly higher phenotypic Axis 1 score suggesting that the APOE locus is not a major risk locus influencing Parkinson’s disease clinical presentation (Additional file 1: Fig S17).
Fig. 4
A high Alzheimer’s disease genetic risk increases the Parkinson’s disease severity risk. a The most influential Axis (Axis 1) is associated with the genetic risk of Alzheimer’s disease The proportion of phenotypic variation explained by the first phenotypic axis derived using these different disease risks (we considered here genome-wide association (GWA) p-value < 0.1) as compared to the original phenotypic axes or exceeding significantly the original phenotypic axis 1 derived using the entire genotype (black horizontal line) or random SNP set respectively (black horizontal line) within Oxford Discovery cohort The colour represent the category of traits: neurodegenerative, neuropsychiatric, metabolic, autoimmune and anthropometric. b The Axis 1 is specifically associated with a biomarker strongly associated with future conversion to dementia In the PPMI cohort, PD patients with higher score for the Phenotypic axis 1 (x-axis), have significant lower CSF level Aβ1–42 (y-axis), a biomarker strongly associated with future conversion to dementia. c The Axis 1 is associated with rapid form of Parkinson’s disease. Boxplot comparing the accuracy of PHENIX to predict the progression of different clinical phenotypes with a general relatedness genetics matrix (blue vs a genetic relatedness matrix calculated by using Alzheimer's disease's genetics variants with GWA p < 0.05 (yellow) in the Oxford Discovery, Tracking UK and PPMI cohort
Our results imply that Parkinson’s disease patients with high genetic risk for Alzheimer’s disease, but excepting APOE, are more likely to develop a more aggressive form of Parkinson’s disease that includes dementia symptoms as indicated by Axis 1, which represents worsening non-tremor, motor phenotypes, anxiety and depression accompanied by a decline in cognitive function (Table 2 and Fig. 2). We tested this hypothesis in the PPMI cohort [10] and found a significant relationship between phenotypic Axis 1 and the cerebrospinal fluid (CSF) Aβ1-42 level (r2 = 0.43, p = 0.007), an Alzheimer’s-associated biomarker strongly associated with future conversion to dementia, but no correlation was observed with total Tau, phosphorylated Tau or Alpha-Synuclein levels (p > 0.05). Parkinson’s disease patients with a high score for phenotypic Axis 1 had a significantly lower CSF level Aβ1–42 [24, 25] (Fig. 4B).
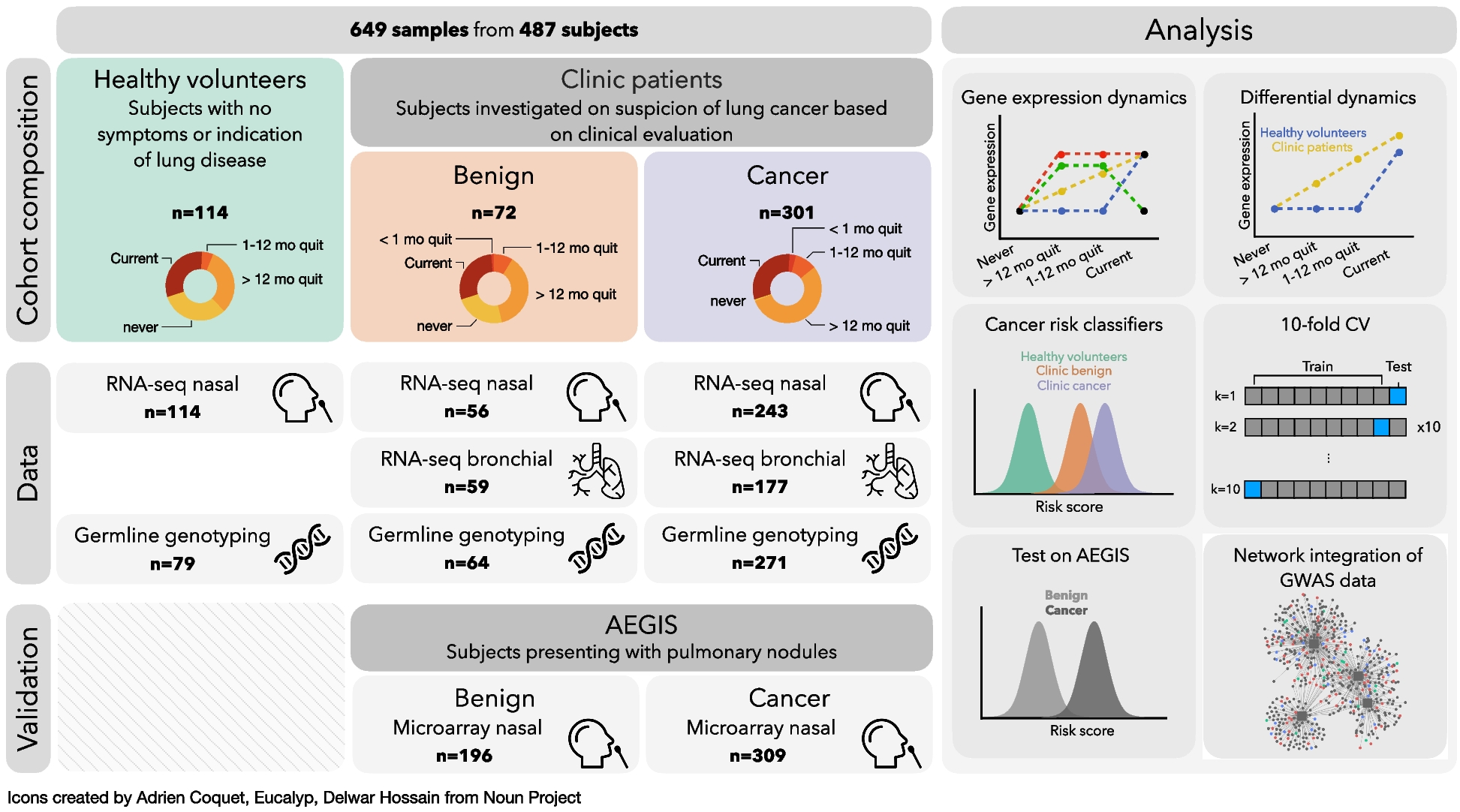
To identify the pathways underlying the genetics of this major clinical axis, we conducted a meta-analysis for the genome-wide association study (GWAS) summary statistic of 4,211,937 variants across 3088 individuals from three cohorts (Oxford Discovery [4, 6], Tracking UK [2] and PPMI [10]) (Method). In line with Alzheimer’s disease risk genetics rather than Parkinson’s disease, we found an association between Phenotypic Axis 1 risk variants and both the SN and cortex microglia-specific genes, which indicates that neuro-inflammation plays a key role in the development of a more aggressive form of Parkinson’s disease (Fig. 4A). Again, following the approach of Agarwal et al. [18] we examined the intersection of genetic risk and microglia-specific functions by identifying highly connected modules of microglia-specific genes whose proteins interacted (Methods) and then used MAGMA to associate these functional modules with different genetic risks. Modules were then annotated with GO terms and corrected for the microglial gene expression background. The genetic risk influencing both Alzheimer’s disease and phenotypic Axis 1 converge to microglia-specific gene Module 2 (Bonferroni adjusted p-value for the number of modules Alzheimer’s disease p = 0.038 – Axis 1 p = 0.042), expressing proteins involved in phagocytosis and regulation of immune response (Fig. 5B).
Fig. 5
The most influential Parkinson’s disease clinical axis involves genetic pathways associated with neuroinflammation. a Identification of substantia nigra (SN) cell types associated with Parkinson’s disease, Alzheimer’s disease, inflammatory bowel disease and ulcerative colitis. To identify the associations between genetic risk variants of different complex traits and cell types SN, we used the MAGMA gene set analysis (one-sided positive two-sample t-test). The heatmap colours give different degrees of significance * and ** indicate nominally significant p (< 0.05) and q value (Bonferroni correction for the number of cell types tested) respectively. b Microglia-specific pathways associated with the genetic risk of the phenotypic Axis 1, Alzheimer’s disease, inflammatory bowel disease (IBD) and ulcerative colitis. Gene Ontology (GO) enrichment for substantia nigra (SN) microglia protein–protein interaction (PPI) genes modules. We tested the convergence of disease genetic risk at a functional level across SN microglia-cell specific PPI gene modules using MAGMA gene set analysis (one-sided positive two-sample t-test); * and ** indicate nominally significant p-value (< 0.05) and q value (Bonferroni correction for the number of PPI modules tested), respectively. The top representative GO biological process (BP) terms are shown for modules. The size of circles represents -log(p) for GO enrichment with Fisher’s test; colours correspond to different modules
As for Alzheimer’s disease, we also found a metabolic influence on the Parkinson’s disease phenotype. We observe that patients with a history of high cholesterol or a history of heart failure, stroke and/or heart attack score significantly higher only on phenotype Axis 1 than those without these histories (Additional file 1: Fig S18). We also observe a significant positive correlation between patient BMI and only their Phenotypic Axis 1 severity score (r = 0.22; p = 3.8e − 06; Additional file 1: Fig S19).
A higher Alzheimer’s genetic risk increases the risk to develop a faster progressing form of Parkinson’sIn the Oxford Discovery [4, 6], Tracking UK [2], and PPMI [10] cohorts, we used their available repeated clinical evaluations to measure individual variation in disease progression. For each clinical phenotype, we derived a progression measure noting that the interval and span of clinical follow-ups varied between the three cohorts (Methods): on average 3.6, 3.6, 11, spanning 4.37, 4.08, and 5.58 years in the Oxford Discovery [4, 6], Tracking UK [2] and PPMI [10] cohort, respectively. The average interval between visits was 20, 19 and 6 months in the Oxford Discovery [4, 6], Tracking UK [2] and PPMI [10] cohort, respectively. We derived the longitudinal phenotypic axes by using the progression measure of each clinical phenotype. We identified a primary axis explaining 76%, 72% and 78% of the clinical progression in the Oxford Discovery [4, 6], Tracking UK [2] and PPMI [10] cohorts, respectively. This axis was firstly correlated with UPDRS III clinical scores for motor symptoms (r2ppmi = 0.83 & r2Tracking = 0.74, r2Discovery = 0.66) and with MOCA scores for the cognitive dysfunctions (r2ppmi = 0.69 & r2Tracking = 0.65, r2Discovery = 0.69). Phenotypically, this axis was significantly similar in three cohorts (Oxford Discovery/PPMI r2 = 0.68, p = 0.04; Oxford Discovery/Tracking UK r2 = 0.90, p = 3 × 10−4, Tracking UK/PPMI r2 = 0.70, p = 0.03) (Additional file 1: Fig S20). A key feature of PHENIX is the ability to impute missing data and thus potentially predict individual disease progression. Using known clinical progression and baseline clinical symptoms of 80% of patients from each cohort, we calculated the accuracy for predicting the progression measure of a clinical phenotype, given the baseline clinical features, by predicting the progression measures in the 20% of patients excluded and repeated this random exclusion and prediction 1000 times (Additional file 1: Fig S21). Accuracy for predicting the progression of cognitive dysfunction was better (Oxford Discovery MOCA test, r2 = 0.42, Tracking UK r2 = 0.40, PPMI r2 = 0.49), than predicting the progression of motor symptoms (UPDRS III Oxford Discovery: r2 = 0.16, Tracking UK: r2 = 0.29, r2Discovery = 0.16). We noted that changes in olfactory function, i.e., changes in Sniffin-16 item odour identification scores in the Oxford Discovery cohort, showed the highest predictive accuracy (r2 = 0.72) when estimating deliberately left-out clinical follow-up measures. In accordance with previous studies, this suggests that hyposmic Parkinson’s disease patients exhibit a worse clinical progression as compared to normosmic patients [26]. Instead of a general genotypic relatedness matrix, using one calculated with only Alzheimer’s disease genetic risk loci significantly improved the accuracy for predicting clinical progression (Oxford Discovery: p = 0.017, Tracking UK: p = 0.003, PPMI: p = 0.04, Fig. 4D). This longitudinal phenotypic axis was significantly correlated with Axis 1 reported above that captures baseline clinical presentations (Oxford Discovery (r2 = 0.31, p < 2.2e−16), Tracking UK (r2 = 0.48, p = 4.12e−14), PPMI (r2 = 0.36, p = 4.12e−14)), indicating that Axis 1 is further associated with rapid progression of multiple clinical symptoms (Additional file 1: Fig S22). All these observations together indicate that genetic risk of Alzheimer’s disease could aid as a prognostic marker for Parkinson’s disease presentation and progression.
留言 (0)