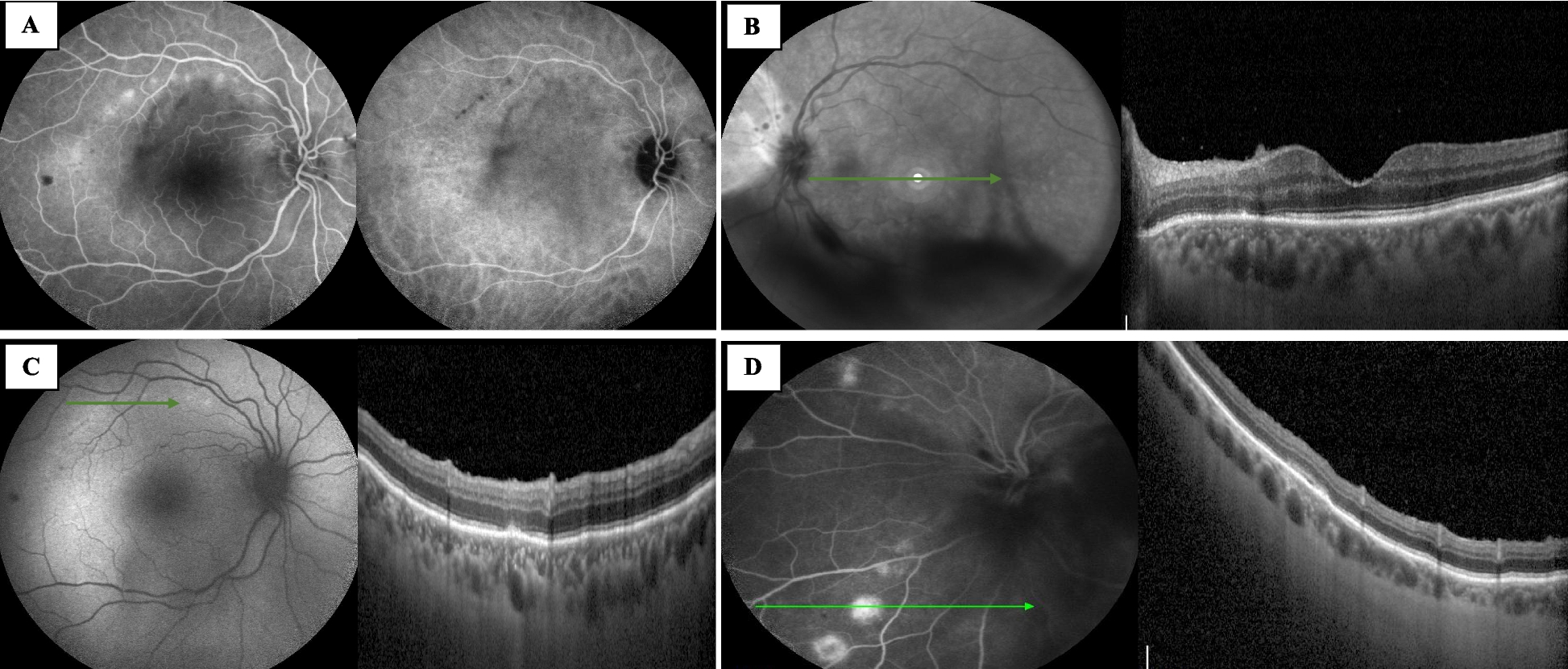
The patient presenting with RP and congenital hearing loss was monitored over a course of 10 years. Progression in RP was observed with an increase in bone spicule deposits, a constriction of the perifoveal hyper-AF ring, and a reduction in ONL thickness. Since PCDH15 is abundantly expressed in both rod and cone photoceptors [6], the patient’s clinical progression indicates significant photoreceptor degeneration associated with the novel two-base-pair deletion (c.60_61del) in PCDH15 (NM_001384140.1). This variant is predicted to produce a premature stop codon in exon 2/33, which is expected to result in nonsense-mediated decay (NMD) of PCDH15 mRNA. The impaired expression of PCDH15 in rod and cone photoreceptors is likely associated with the patient’s progression in RP with a loss in rod photoreceptors and secondary cone degeneration. Although the exact molecular function of retinal protocadherin-15 is ambiguous [7], using existing animal models with PCDH15 variants may help further understand the patient’s clinical observations. A similar founder variant in PCDH15 that is unique to Ashkenazi Jews, p.Arg245Ter, was simulated using a Pcdh15R250X knockin mutant mouse model that phenocopied human p.Arg245Ter congenital hearing loss and abnormal vestibular function [7]. While present in wild-type mice, immunostaining revealed the absence of protocadherin-15 expression in the inner segments of photoreceptors, outer plexiform layer, the ganglion cell layer, and retinal pigment epithelium (RPE) in Pcdh15R250X mice. Under photopic conditions, the loss of protocadherin-15 hinders the transportation of arrestin and transducin between the photoreceptor outer segment (OS) and inner segment (IS) to desensitize or bind to opsin, respectively. This results in abnormal protein localization in the phototransduction cascade and retinoid cycle [7]. Additionally, a reduction in enzymes CRALBP and RPE65 consequently reduced 11-cis-retinal functions [7]. Combined with the gross retinal degeneration observed in the patient, both observations may help explain extinct ffERG amplitudes in Supplementary Fig. 1. However, acute retinal degeneration was less severe in mice when compared to human pathophysiology [7]. This may be attributed to the absence of protocadherin-15 associated calyceal processes in rodent photoreceptor cells that are present in humans, frogs, and monkeys [8]. Knockdown PCDH15 frog models show the degeneration and loss of photoreceptor function due to the proposed role of the calyceal process in rod and cone maintenance and development [8].
The c.60_61del variant was identified as homozygous. Although the parents were not known to be related to each other, the patient had regions of homozygosity (ROH) across ~ 0.8% of the genome suggesting that the parents are distantly related. The PCDH15 homozygous variant was found within one of the larger ROH (~ 10 Mb). Therefore, even though the parents were not available for Sanger sequencing, it is likely that each parent is heterozygous for the variant. There was no evidence of a large copy-number-loss variant spanning the PCDH15 gene from the WES data.
Loss of function variants are known to be the disease-causing mechanism with many pathogenic variants reported in the literature and databases being null variants. Clinical observations reported a loss of function in the patient’s phenotype, which may be attributed to NMD mechanisms eliminating mRNA containing a premature termination codon [9]. Since the variant is located at the beginning of the PCDH15 gene on exon 2/33, nonsense-mediated decay of PCDH15 mRNA is expected to occur. However, if a small amount of mRNA does escape the NMD pathway, then the shortened peptide (around 21 amino acids), will most likely be degraded. Hence, it is likely that no protein products are produced. To further examine the mechanisms that relate to a loss of function, future animal models may use qPCR techniques to determine the expression of PCDH15 mRNA to examine the role of NMD in this novel variant.
In this longitudinal report, we followed a patient with a novel variant in the PCDH15 gene over a course of 10 years. A novel nonsense variant c.60_61del results in typical USH1F clinical symptoms, such as congenital hearing loss and progressive RP [2]. Although the patient was first examined in her twenties, with clinical features resembling clinical subtypes of USH 2 and USH 3 [2], her ERG responses (Supplementary Fig. 1) resemble RP in the advanced stage [10] suggesting a prepubertal onset of photoreceptor degeneration, which correspond with bilateral ONL thinning in SD-OCT (Fig. 2d). Thus, ffERG is essential for an early and correct diagnosis of USH1 when combined with the presence of congenital hearing loss, allowing clinicians to test genes related to USH1.




留言 (0)