
記住我
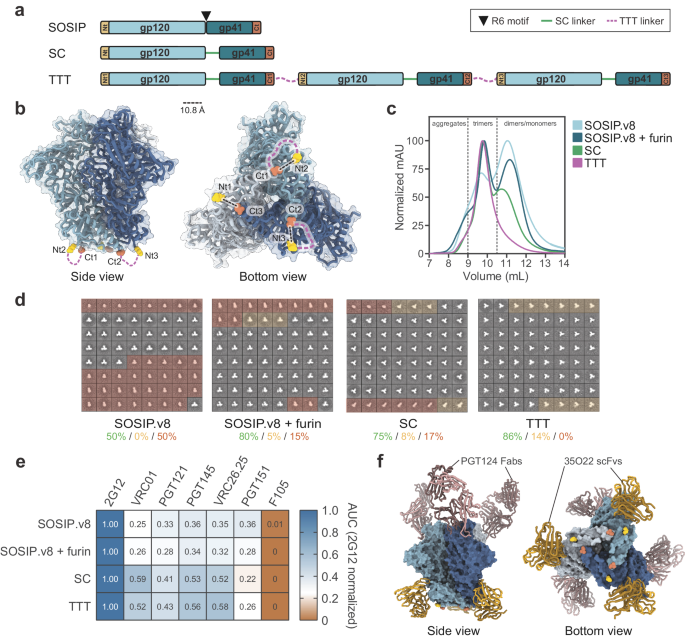
Replication-incompetent, E1/E3-deleted, recombinant Ad26 and Ad35 vectors were engineered using the AdVac® system (Janssen)11,45, as described in detail for Ad26.ZEBOV, Ad35.ZEBOV, Ad26.SUDV, and Ad26.Mos4.HIV7,24,46. Ad26.Mos4.HIV consists of 4 Ad26 vectors: Ad26.Mos.1.Env (encodes a mosaic insert of the Env protein sequence), Ad26.Mos2S.Env (encodes modified Mos2 HIV-1 Env protein sequence), Ad26.Mos1.Gag-Pol (encodes Mos1, HIV-1 Gag and Pol protein), and Ad26.Mos2.Gag-Pol (encodes Mos2 HIV-1 Gag and Pol protein). Briefly, codon-optimized genes encoding the relevant transgene were inserted into the E1 position of the Ad genomes under transcriptional control of the human cytomegalovirus promoter and the SV-40 polyadenylation sequence.
Cloning, rescuing, and manufacturing of the replication-deficient Ad vectors using the complementing cell line PER.C6® (Janssen)45. Viral particles (vp) titers in the viral preparations were quantified by measurement of optical density at 260 nm47, and infectivity (expressed as infectious units [IU]) was assessed by tissue culture infective dose 50%48,49. The vp/IU ratio was between 1:1 and 6:1 for the viral preparations. Ad-mediated expression of the various transgenes was confirmed by Western blot analysis of cell culture lysates from infected A549 cells or by polymerase chain reaction.
MVA vectorsMVA-BN-Filo is a trivalent recombinant MVA strain Bavarian Nordic (MVA-BN)–based filovirus vaccine directed against Marburg virus and Ebola virus infection. The full-length coding sequences for GP antigens of MARV Musoke, EBOV Mayinga, and SUDV Gulu, as well as the nucleoprotein from Taï Forest Ebola virus, were codon optimized, synthesized (GeneArt, Regensburg, Germany), inserted into MVA-BN46. MVA-mBN414A compromising a single MVA-BN vector was genetically engineered to encode Mos1.Env, Mos2S.Env, Mos1.Gag-Pol, and Mos2.Gag-Pol HIV-1 protein sequences17. MVA-RSV.FA2 (MVA-mBN235A) is a monovalent vaccine comprising a single MVA-BN vector genetically engineered to encode the F fusion protein of RSV strain A2 under the synthetic early-late promoter PrS. Primary chicken embryo fibroblast cells used for recombinant live, attenuated MVA viral vector–based vaccine generation and production were prepared from embryonated eggs and maintained in serum-free conditions.
Animals and housingFor study 1, a total of nineteen 5- to 6-year-old, healthy female cynomolgus macaques (Macaca fascicularis) of Vietnamese origin (body weight 3–8 kg at study start) were rolled over from a previous study9. The animals were originally purchased from Covance (Alice, TX, USA). Five of the animals had received previous vaccination with an Ad26/Ad26 homologous regimen, and 5 had previously received an Ad26/Ad35 heterologous regimen; the insert encoded by these vectors was RSV.FA29. Nine other animals were vaccine-naïve upon enrollment.
For study 2, twelve 4- to 5-year-old, healthy female cynomolgus macaques of Mauritian origin (body weight 3–7 kg at study start) were rolled over from a previous study and purchased by Charles River Edinburg. Six of the animals had previously received 2 doses of an Ad26 vector expressing a fusion protein of RSV.FA2 and Gaussian luciferase and a dose of MVA encoding RSV.FA2, and 6 were vaccine-naïve upon enrollment.
For study 3, twelve 4- to 7-year-old, healthy male and female cynomolgus macaques of Chinese origin (body weight 3–7 kg at study start) were rolled over from a previous study and purchased by Charles River Reno NV and Alpha Genesis Inc. Six of the animals had previously received an MVA-Ad26 heterologous regimen; the inserts of those vectors were EBOV GP for Ad26 and MVA-BN-Filo. Six other animals were vaccine-naïve upon enrollment.
All animals were kept in a biosafety level 2 facility under specific pathogen-free conditions after screening negative for Mycobacterium tuberculosis, simian immunodeficiency virus, simian retrovirus, and simian T-lymphotropic virus. Screening included herpes B virus and measles serology.
Animals in study 1 and study 3 were pair-housed in groups of 2 or 3 animals in stainless steel cages placed in study-dedicated, USDA- and OLAW-approved rooms, while animals in study 2 were socially housed in groups of 6 in 2-story gang pens, which were also OLAW approved. Animals in all 3 studies were kept under controlled, recorded, environmental conditions of humidity, temperature, and light (12-h light cycle). For all 3 studies, animals of the same sex and study group per cage were co-housed, except for brief, procedure-related periods. Animals were provided with sensory and cognitive environmental enrichment, including manipulatable objects and foraging devices. Three times a day, animals were fed a standard NHP diet consisting mainly of high-protein monkey biscuits but also including PRIMA-Treats®, a soft dough diet, and a selection of fresh fruit, peanuts, cereals, or other treats. Tap water was provided ad libitum through an automated system. Animal well-being was monitored daily by husbandry staff, and routine animal health surveillance, including evaluation of blood chemistry and hematology, was provided by veterinary staff. Pre-set humane endpoints were used by a veterinarian to define study-unrelated sacrifice criteria. All measures were taken to minimize pain, distress, and suffering, and all procedures were performed by trained personnel.
Study design and animal proceduresFor study 1 and study 3, all animal procedures were performed under anesthesia either with ketamine (10–15 mg/kg intramuscularly) or Domitor (0.015 mg/kg intramuscularly). For study 2, animals were trained; therefore, vaccination and blood sampling were performed without the use of anesthesia.
Animals were assigned to the study treatment groups based on the A-series vaccine administered, receiving a vector backbone–homologous regimen matching the initial regimen for the B-series (e.g., an Ad26/Ad26 regimen twice). The initial vaccinations were given at either 12-week (studies 1 and 2) or 8-week (study 3) intervals. Treatment groups in the present study were named according to the administered vector backbones, with “rep” indicating the sequence was identical to that received previously. The interval between the last dose of the first study (A-series) and the first dose of the subsequent vaccination (B-series) was 55 weeks for study 1, 26 weeks for study 2, and 57 weeks for study 3 (Fig. 1).
Animals in study 1 were divided into 4 study groups, with 4 to 5 animals per group (Supplementary Fig. 1). The animals received 2 doses (5 × 1010 vp) of either Ad26.ZEBOV (2-dose homologous regimen, Ad26/Ad26 rep group) or Ad26.ZEBOV followed by Ad35.ZEBOV (2-dose heterologous regimen, Ad26/Ad35 rep group), with an 8-week interval between doses. Control animals who had not received any prior treatment received either a 2-dose homologous Ad26/Ad26 regimen (Ad26/Ad26 group) or a 2-dose heterologous Ad26/Ad35 regimen (Ad26/Ad35 group).
Animals in study 2 were divided into 2 study groups (pre-exposed or unexposed), with 6 animals per group (Supplementary Fig. 1). Both groups received a 2-dose heterologous vaccination regimen of 5 × 1010 vp of Ad26.SUDV at dose 1, followed 8 weeks later by 108 vp units (IFU) of MVA-BN-Filo (Ad26/MVA rep group and Ad26/MVA group).
Animals in study 3 were divided into 2 study groups (pre-exposed or unexposed), with 6 animals per group (Supplementary Fig. 1). Both groups received a 2-dose heterologous vaccination regimen of 5 × 1010 vp of Ad26.Mos4.HIV, followed 12 weeks later by 108 vp units (IFU) of MVA-mBN414A (Ad26/MVA rep group and Ad26/MVA group).
All vaccines were administered in a 0.5-mL volume intramuscularly in the quadriceps with the indicated vector particle-doses in formulation buffer. Venous blood for PBMC isolation or serum was collected from the femoral vein. Blood volumes taken did not exceed 12 mL/kg within 30 days, and a maximum of 9 mL/kg at each individual bleeding time point.
Processing of peripheral bloodSerum samples were prepared from clotted blood drawn into serum tubes after spinning at 1900 G for 5 min at room temperature (RT). Serum was stored at –80 °C until time of analysis. PBMCs were isolated from whole blood drawn into anticoagulant-containing tubes (ethylenediaminetetraacetic acid [EDTA]) by Ficoll density gradient centrifugation. Blood was diluted 1:1 with Dulbecco’s phosphate-buffered saline (D-PBS) without Ca2+ or Mg2 + (Quality Biological, Gaithersburg, MD, USA), underlayed with an equal volume of Ficoll-Paque Plus (GE Healthcare, Little Chalfont, UK), spun at 1750 G for 40 min at RT. Buffer layers were transferred into a fresh tube, washed 3 times with D-PBS, and spun at 393 G for 5 min at RT. When needed, lysis of residual red blood cells (RBCs) in RBC Lysis Solution (Qiagen, Hilden, Germany) or ACK lysis buffer (Lonza Bio Whittaker) for 10 to 15 min at RT was performed. Lysis was stopped by addition of excess D-PBS, and tubes spun at 1750 G for 5 min at RT. Viable cell numbers were subsequently determined by trypan blue exclusion using a Countess Automated Cell Counter (Thermo Fisher Scientific, Hampton, NH, USA) or using ViaCount reagent with a third-generation Guava® Easycyte™ cytometer (Luminex, Austin, TX, USA). For cells processed directly for IFNγ ELISpot, cells were adjusted to a concentration of 2 × 106 cells/mL in RPMI-10 (RPMI complemented with 10% fetal bovine serum [FBS]; Thermo Fisher Scientific), 10 mM HEPES buffer (Quality Biological), 2 mM L-glutamine (Quality Biological), and 100 ug/mL penicillin/streptomycin (Quality Biological) and kept on ice until analysis. Alternatively, cells were adjusted to a concentration of 5 × 106 cells/mL in CryoStor® (Biolife Solutions, Bothell, WA, USA), and samples were aliquoted and transferred into liquid nitrogen until use.
Frozen cells were thawed in a 37 °C water bath, washed with RPMI-10, spun at 400 G for 5 min at RT, and subsequently washed twice. Cells were counted as indicated previously using ViaCount reagent with a Guava® cytometer. Cells were adjusted to a concentration of 107/mL and cultured in T-25 flasks in a 37 °C, water-jacked 5% CO2 incubator for 18 to 24 h. Following incubation, cells were counted using ViaCount reagent with a Guava® cytometer, adjusted to a concentration of 2.5 × 106 cells/mL in RPMI-10, and kept on ice until analysis.
Adenoviral neutralization assayAd26 Nab titers in serum were assessed using a luciferase-based virus neutralization assay (VNA)50. Briefly, heat-inactivated Cynomolgus Macaque serum samples was 2-fold serial diluted starting at a 1:32, 1:64 or 1:128 dilution for Ad26 (depending on the study, see figure legends for the exact start dilution). E1/E3 deleted Ad26-luciferase reported constructs were combined with serial diluted serum into a Tissue Culture treated Black and White Isoplate-96 (Perkin Elmer, Nederland B.V) at 500 to 1000 vp/cell. Plates were incubated for 30 minutes at RT, before A549 human lung carcinoma cells (ATCC® CCL-185™, American Type Culture Collection, Manassas, VA, USA) were added at a density of 1×104 cells/well. After incubation for 20 to 24 hours at 37 °C and 10% CO2, luciferase activity was measured using the Neo-Lite Luciferase Assay System (Perkin Elmer, Waltham, MA, USA) and a BioTek Synergy Neo luminescence counter (BioTek/Agilent, Santa Clara, CA, USA) or EnVision multimode plate reader (Perkin Elmer). A 90% neutralization titer (IC90) was defined as the maximum serum dilution that neutralized 90% of luciferase activity. Each serum sample was analyzed in duplicate.
IFNγ ELISpotAntigen-specific, IFNγ-secreting T cells were enumerated in isolated PBMCs using an ELISpot kit specific for monkey IFNγ (Monkey IFNγ ELISpotPRO; MabTech, Nacka Strand, Sweden). Plates pre-coated with an NHP IFNγ-specific capture antibody (clones GZ-4 or MT126L) were washed 4 times with sterile D-PBS (180 µL/well) and blocked with RPMI-10 (200 µL/well) for 30 min at 37 °C and 5% CO2. After removal of the blocking buffer, PBMCs in RPMI-10 were seeded at 2 to 5 × 105 cells/well and stimulated with peptide pools reconstituted in dimethylsulfoxide (DMSO), consisting of 15-mers overlapping by 11 amino acids at a final concentration of 2 µg/mL for 18 to 20 h at 37 °C and 5% CO2, in a final volume of 200 µL.
For study 1, 2 peptide pools covering the EBOV GP protein N-terminal and C-terminal sequences were used to limit the number of peptides per pool (43–58 peptides/pool). The results of the N-terminal and C-terminal pools for EBOV GP were pooled for reporting purposes.
In addition, 5 peptide pools containing shared and specific peptides for Ad26 and Ad35 hexon protein were used. Responses are reported as either Ad26-hexon specific (sum of 2 Ad26-specific peptide pools) or pan-Adeno Hexon response (sum of the responses to the 5 peptide pools).
For study 2, 2 peptide pools covering the SUDV Gulu GP protein N-terminal and C-terminal sequences were used to limit the number of peptides per pool (43–58 peptides/pool). Peptides that overlapped with more than 9 consecutive amino acids within the EBOV Mayinga, SUDV Gulu, and TAFV strains or MARV Angola and Ravn strains were combined into a consensus pool, SUDVcon (~100 peptides/pool). The responses given in the figures are a sum of the background-subtracted responses induced to the 3 peptide pools (N-terminal and C-terminal pools and SUDVcon for SUDV GP).
For study 3, individual peptide pools covering the Env (Env-1, Env-2, and Env-3), the Gag (Gag-1 and Gag-2), the Pol (Pol-A, Pol-B, and Pol-C) were used25. The total Env response given in the figures is a sum of the background-subtracted response induced to the 3 individual sub-Env pools (Env-1, Env-2, and Env3). Likewise, the total Gag and total Pol response is a sum of the responses elicited to the individual sub-Gag pools and sub-Pol pools after background subtraction, respectively.
RPMI-10 supplemented with 0.005% to 0.33% DMSO served as a medium control and a 1/1000 dilution of α-CD3 antibody or 5.5 mg/mL phytohemagglutinin in RPMI-10 served as a positive control. After removal of the cell suspension, wells were washed 5 times with PBS + at RT and subsequently incubated with IFNγ-detector antibody conjugated to alkaline phosphatase (clone 7-B6-1-ALP, 1:200 in PBS + 0.5% FBS) for 2 h at RT. Plates were washed 5 times with PBS + at RT, and spots developed for 15 min in the dark at RT using a 5-bromo-4-chloro-3’-indolyphosphate p-toluidine/nitro-blue tetrazolium chloride solution filtered through a 0.45-µm filter. The development was stopped by washing extensively with tap water. Plates were air dried for at least 24 h before spots were counted on an ImmunoSpot S5 ELISpot plate reader (C.T.L. Europe GmbH, Bonn, Germany) or A·EL·VIS ELISpot plate reader (A·EL·VIS, Hannover, Germany), and counting was done with Eli·Analyse ELISpot Image Analysis software (A·EL·VIS). All samples were analyzed in either duplicate or triplicate. Mean SFU/106 cells were calculated from the replicate measurements, followed by individual background subtraction of the mean medium control values from the mean peptide-stimulated values. For antigens covered by >1 peptide pool, background-subtracted mean peptide–stimulated values were summed per animal per time point. Based on historical data, the background/threshold was empirically set at 50 SFU/106 PBMC. Values below the threshold of 50 SFU/106 PBMC were set at half that threshold (25 SFU/106 PBMC) for the purpose of graphical representation. For calculation of the fold-change, values below the threshold of 50 SFU/106 PBMC were set at this threshold. To calculate the fold-change from pre–dose 1 to peak response post–dose 1, the peak response post–dose 1 per animal was divided by the response measured pre-dosing (all studies, week –2). Similarly, to determine the fold-change from pre–dose 2 to peak response post–dose 2, the peak response post–dose 2 per animal was divided by the response measured pre-dosing (study 1, week 8; study 2, week 4; study 3, week 12).
Determination of EBOV GP–specific IgG in serum by ELISATotal serum IgG targeting GP of EBOV was assessed by an ELISA that was qualified and validated for human sera46. Briefly, Maxisorp™ 96-well plates (Nunc-Immuno) were coated overnight at 4 °C with Galanthus Nivalis Lectin (GNA; Sigma-Aldrich, Burlington, MA, USA) diluted in PBS at 10 μg/mL. Remaining lectin solution was removed and 200 μL PBS/10% FBS added for 90 min at RT. The plates were washed 5 times with PBS/0.2% Tween20 (PFS-T; Sigma-Aldrich) at RT, coated with supernatant-containing recombinant filovirus GP for 90 min at RT, and then washed again. Serum from NHPs was serially diluted (starting dilution, 1:50) in sample buffer (PBS/0.2% Tween/1% FBS). 100 μL of diluted sample was transferred to the coated Maxisorp™ 96-well ELISA plates, incubated for 90 min at RT, and washed 5 times with PBS/0.2% Tween20 at RT. Bound IgG was detected with goat–anti-human IgG (H + L) conjugated to horseradish peroxidase (MilliporeSigma, Burlington, MA, USA). The substrate Sigma fast o-Phenylenediamine dihydrochloride (OPD, Sigma-Aldrich, Burlington, MA, USA) was added for 10 min at RT. The enzymatic reaction was stopped by addition of 3 M H2SO4 and measured at 492 nm. IC50 values were calculated by 4-parameter curve-fit and compared against a filovirus GP strain-specific reference serum and expressed as ELISA units (EU) /mL.
Determination of SUDV GP–specific IgG in serum by ELISATotal serum IgG targeting GP of SUDV Gulu was determined by ELISA. Maxisorp™ 96-well plates were coated overnight at 4 °C with purified SUDV GP protein (produced internally at Janssen Vaccine & Prevention) diluted in 20 mM Tris-HCl solution at a concentration of 0.25 μg/mL. After washing 3 times with 200 μL PBS-T at RT, plates were blocked with 180 μL PBS/10% FBS at RT for 90 min. The plates were washed 3 times as indicated previously with PBS-T. NHP serum was serially diluted (3-fold steps) in sample buffer starting at a dilution of 1:45 (PBS/0.2% Tween/1% FBS, sample buffer) in round-bottom polypropylene plates (Nunc Cat#267245).
100 μL of diluted sample was transferred to Maxisorp™ 96-well ELISA plate and incubated at RT for 60 min. Plates were washed 3 times with PBS-T as indicated previously. Bound IgG was detected with goat–anti-human IgG (H + L) conjugated to horseradish peroxidase (MilliporeSigma, Burlington, MA, USA), diluted 1:8000 in sample buffer and incubated for 1 h at RT. Plates were washed 3 times with 200 μL PBS-T. 100 μL of Sigma Fast OPD solution (Sigma-Aldrich, Burlington, MA, USA) was added and incubated for 10 min at RT. The reaction was stopped using H2SO4 and measured at 492 nm. Endpoint concentrations were compared against a filovirus GP strain–specific reference serum and expressed as EU/mL.
Determination of Env Clade C and Mos1-specific IgG in serum by ELISAAntibody binding to the Clade C gp140 and mosaic gp140 antigens was determined by ELISA51. Briefly, antigen (HIV_Env_C_C97ZA and HIV_Env_Mos124) was coated at 1 μg/mL in PBS and serum samples were tested undiluted, resulting in 1/10 serum dilution in the final ELISA plate, and incubated on plates. Binding antibody was determined using horseradish peroxidase-conjugated detection antibody mouse–anti-human IgG (Jackson Cat#209-035-011, 1:20,000) and SureBlue TMB (SeraCare 5120-0047; Kirkegaard & Perry Laboratories). The final concentration of each sample was calculated using Gen5 software (BioTek/Agilent). The concentration is equivalent to the back-calculated concentration of the measured OD450 value onto the 4PL curve-fit of the standard curve24.
EBOV pseudovirus neutralization assaysThe filovirus pseudovirion VNA was performed as follows25, and pseudovirus preparations were generated by co-transfection of human embryonic kidney (HEK) 293 cell cultures, with a replication-defective retroviral vector containing a luciferase gene along with an expression vector containing EBOV Makona GP sequence. Pseudovirus stocks were generated and characterized for suitability to assess EBOV-specific neutralization. Pseudoviruses were incubated with serial dilutions of serum samples and used to infect HEK293 cell cultures. Each serum sample was serially diluted 10 times (4-fold), starting from a dilution of 1:40. The ability of serum to neutralize EBOV pseudovirus infectivity was assessed by measuring luciferase activity ~72 h post–viral inoculation versus a control infection using a murine leukemia virus envelope–pseudotyped virus. IC50 values were expressed as the reciprocal of the serum dilution that inhibited the virus infection by 50%.
RSV A2 Virus neutralization assayVNA against RSV A2 was determined on serum of animals from study 2 using recombinant luciferase expressing RSV viruses9. Five thousand A549 human lung carcinoma cells were added to each well of 96-well white half-area plates (Greiner Bio-One, Frickenhausen, Germany) containing 2.5 × 104 SFU/well of RSV-A2 viral particles encoding a luciferase reporter gene (resulting in a multiplicity of infection of 5), together with serial dilutions of individual heat-inactivated cynomolgus macaque serum. After incubation for 20 h at 37 °C and 10% CO2, luciferase activity in lysed cells was measured using the Neo-Lite Luciferase Assay System (Perkin Elmer) on a BioTek Synergy Neo luminescence counter (BioTek/Agilent). IC50 values were defined as the maximum serum dilution that neutralized luciferase activity by 50%. Each serum sample was analyzed in duplicate.
Statistical analysesImmunologic parameters (i.e., ELISA, VNA, ELISpot) were log-transformed. Two types of analysis were performed: (1) between group comparisons per time point per study using ANOVA for potentially censored values (Tobit model) and (2) within group comparisons of fold-changes over time using ANOVA. The fold-changes in response after the A-series and B-series vaccination (i.e., pre–dose 1 vs post–dose 1 and pre–dose 2 vs post–dose 2) for all immunologic parameters (i.e., ELISA, VNA, ELISpot) were calculated per animal and then log-transformed. For study 3, in addition to the comparisons per antigen, the average fold-changes over the Mos1 and Clade C antigens (ELISA) and over the Gag, Pol, Env antigens (ELISpot) were calculated and analyzed. Vaccine regimens were subsequently compared using ANOVA, both per study and pooled across studies. P < 0.05 were considered statistically significant, and a Bonferroni correction for 2 comparisons was applied for the analysis of study 1.
Reporting summaryFurther information on research design is available in the Nature Research Reporting Summary linked to this article.
留言 (0)