
記住我


Ischemic stroke (IS) is an acute cerebrovascular disease caused by brain tissue damage resulting from the sudden rupture of blood vessels in the brain or blocked blood vessels flowing into the brain. Under the condition of ischemia and hypoxia, nerve cells produce a series of biochemical cascade reactions leading to irreversible brain damage. Stroke results in substantial social and economic burdens around the world. Globally, age-standardized stroke mortality decreased between 1990 and 2010, but age-standardized stroke incidence did not change significantly, and the absolute number of strokes, stroke survivors, and related deaths each year continued to increase [1]. However, as a result of the lack of understanding surrounding the mechanisms involved in ischemic brain injury, there is no specific clinical drug in use. A growing body of evidence suggests that genetic factors can be used to explore new drug targets and treatments for stroke, and even to improve stroke diagnosis and preidentification of those at risk [2].
Sequences longer than 200 nucleotides that do not encode proteins are called lncRNA and can function independently as transcripts [3]. Although lncRNA was initially regarded as simple transcription ‘noise’ by researchers, subsequent studies show that lncRNA participates in various pathophysiological situations, such as apoptosis, cycle progression, differentiation, and inflammation, by regulating the stability and nuclear retention of target genes [4]. LncRNAs normally interact with one or more RNA-binding proteins (RBPs) to perform a variety of cellular functions. The majority of these processes involve various information carriers other than mRNA to exert biological effects, for example, they may utilize splicing, polyadenylation, transport, stability, and translation [3]. Studies have shown that lncRNA whose expression level changes during cerebral IS can regulate gene expression at the transcriptional and posttranscriptional level; thus, it may have potential as a biomarker and therapeutic target [5].
Taurine upregulated gene 1 (TUG1) is observed when genomic screening for upregulated genes after taurine treatment and is highly expressed in the mammalian brain. TUG1 is a key signal molecule in the development of the retina, and its deletion seriously damages the formation of mouse retinae [6]. In clinical trials focusing on the correlation between TUG1 polymorphism and the risk of IS, a more effective overexpression promoter may be combined with globin transcription factor 1 (GATA-1) to increase the level of TUG1, demonstrating that TUG1 may represent an independent risk of IS [7]. At present, studies regarding the proapoptotic effect of TUG1 in ischemia–hypoxia experiments almost exclusively focus on the competing endogenous RNA mechanism of LncRNA. However, TUG1’s potential role and molecular mechanism in promoting neuronal apoptosis in cerebral ischemia need to be further explored.
The RBP, human antigen R (HuR, also known as ELAV), regulates proliferation, senescence, differentiation, apoptosis, and stress and immune responses by controlling the splicing, localization, stability, and translation of intracellular transcripts, including coding and noncoding transcripts [8]. Among the different proteins that specifically bind to AU-rich elements (AREs), members of the ELAV protein family, especially the ELAV-like protein HuR (HuA), regulate the half-life and stability of target mRNAs by binding to U- or Au-rich regions [9]. As one of the most well-studied RBPs, HuR binds to the 3′-untranslated region (UTR) transcripts of mRNAs, including p53, p27, Caspase-9, and BCL2 [10]. The HuR protein is mainly located in the nucleus, and it performs its mRNA stabilizing function by shuttling between the nucleus and the cytoplasm [9]. Therefore, the cytoplasmic role of HuR in neuronal cells under ischemia may be vital in terms of deciphering the mechanism of apoptosis in diseased cells. Moreover, the effect of specific lncRNAs on nucleocytoplasmic HuR shuttling in this state has not been described.
In this study, we hypothesized that lncRNA and RBP directly bind to each other in the nucleus and regulate the subcellular spatial positioning of RBP, enabling it to play its role as a key mediator of apoptosis. In order to verify this hypothesis, we knocked down TUG1 in the nucleus and cytoplasm separately to observe the changes in the subcellular localization of RNA-binding proteins. These findings may provide insight into lncRNA TUG1 as an important regulator of the pathophysiology of mouse hippocampal neuronal cell line (HT22) cell necrosis through direct interaction with the RNA-binding protein HuR (a key mediator of apoptosis).
Methods Cell cultures and transfectionHT22 was purchased from Procell, Wuhan, China. The company recommended the ‘HT22 cell special medium’ (Procell, Wuhan, China, CM-0697), which contains DMEM (Procell, Wuhan, China, PM150210), 10% fetal bovine serum (FBS) (Procell, Wuhan, China, 164210-500), and 1% P/S (Procell, Wuhan, China, PB180120). The cell gas phase culture conditions were as follows: air: 95%; CO2: 5%; temperature: in an incubator at 37 °C. When the cell confluence reached 80%, the cells were passaged at a ratio of 1:6. Generally, the medium was changed, or the cells were passaged after about 2 days.
According to the instructions of the Lipofectamine 2000 Reagent transfection kit (Invitrogen, California, USA 11668-027), the product was mixed with siRNA-TUG1 (Sangon, Shanghai, China) or antisense oligonucleotides (ASO)-TUG1 (Sangon, Shanghai, China) in order to transfect HT22, and the transfection efficiency was assessed using qPCR 24 h after transfection.
Oxygen-glucose deprivation modelThe medium was aspirated in the HT22 cell culture dish and washed with PBS several times. Then, an equal volume of glucose-free DMEM (Procell, PM150270) was added, and it was placed in a tri-gas incubator (Heraeus, Hanau, Germany) at 37 °C, 0.5% O2, 94.5% N2, and 5% CO2 with oxygen-glucose deprivation (OGD) culture. Thereafter, the experimental group removed the sugar-free DMEM and washed it with PBS before proceeding to the next step of the experiment. The same conditions were used for the control group, except that it was not exposed to OGD and the sugar-free DMEM was replaced with complete medium.
Protein isolation and Western blot analysisIn order to measure the relative content of HuR protein in different samples, we strictly followed the manufacturer’s instructions using Western and IP cell lysis buffer (Beyotime, Nanjing, China, P0013) to extract total protein from cell samples, and a Nuclear and Cytoplasmic Protein Extraction Kit (Beyotime, Nanjing, China, P0027) to extract and separate proteins from the cytoplasm and nucleus. Herein, we used a BCA Protein Assay Kit (Beyotime, Nanjing, China, P0012) for protein quantitative determination according to the instructions. The same amount of protein sample was electrophoresed in 12% SDS-PAGE, and then transferred to a PVDF membrane. After blocking with a TBS-T (NCM Biotech, Nanjing, China, WB21000) solution containing 5% skimmed milk powder (Servicebio, Wuhan, China, G5002) at room temperature for 1 h, the membrane was completely immersed in the primary antibody, diluted at 1:1000 in the primary antibody diluent, and incubated overnight at 4 °C. Anti-β-actin (ABclonal, AC026) and HuR (ABclonal, A19622) are both Rabbit mAbs. The membranes were washed with TBS-T three times for 10 min each, and then the membranes were incubated with the secondary antibody for 2 h at room temperature. Subsequently, the excess secondary antibody was removed by TBS-T washing three times for 10 min, and then the enhanced chemiluminescence detection kit (catalog number 32209; Thermo Fisher Scientific, Inc.) was used with a chemiluminescence system [Tanon Chemidoc Apparatus (Tanon-Bio, Shanghai, China 2500)] to observe the blot.
Cholecystokinin-octopeptide-8 assayIn order to determine cell viability, 8000 cells per mixed with 100 ul of complete medium and seeded in 96-well plates. Before measuring the absorbance of cells at 450 nm using a microplate reader, the original medium was removed and the cells in each well were washed with PBS. Then, the cell HT22 cell special medium (Procell, CM-0697) containing 10% Cell Counting Kit-8 (Vazymy, Nanjing, China, A311-01) was mixed in a 15-ml tube and added to a 96-well plate at 100 ul per well. The product was then placed in a 37 °C cell incubator for 1 h, according to the manufacturer’s instructions. Each treatment was performed in at least six replicate wells.
Crosslink cellsThe speed was set to 1000 rpm and the mixture was spun gently for 4 min to collect the cells. Then, the cells were resuspended in room-temperature phosphate-buffered saline (PBS, Servicebio, G4202), and the cells were washed to 5 mol/l cells/ml, before being centrifuged at 2000 rpm for 2 min. The cells were immediately resuspended in DMEM (Servicebio, G4510) medium without fetal bovine serum (FBS) or P/S to 5 mol/l cells/ml at room temperature, formaldehyde was added without methanol to a final concentration of 0.1%, and the mixture was spun slowly at room temperature for 5 min. Then, we added filtered glycine to a final concentration of 125 mmol/l, centrifuged it at 2000 rpm for 2 min, and then washed it twice with PBS and PIC at a concentration of 10 mol/l cells/ml at 4 °C. Finally, the mixture was rotated at 700 rpm for 8 min at 4 °C before being rotated at 2000 rpm for 2 min. Then, the supernatant was removed and the cell pellet was quick-frozen in liquid nitrogen and stored at −80 °C.
Formaldehyde RNA immunoprecipitationIn order to determine the interaction between TUG1 and HuR protein, we used HT22 cells to perform the formaldehyde RNA immunoprecipitation (fRIP) experiment for RNA-binding protein HuR. We modified the existing RNA IP (RIP) and chromatin immunoprecipitation (ChIP) protocols to optimize RNA and protein recovery. We combined 0.5 ul/ml 1 mol/l dithiothreitol (DTT), 10 ul/ml Protease Inhibitor Mix (Thermo Scientific, Massachusetts, USA, PI-87785), and 2.5 ul/ml RNaseOUT (Thermo Scientific, Massachusetts, USA) with 1 ml RIPA lysis buffer (Servicebio, G2002) per 10 mol/l cells, resuspended the crosslinked cryoprecipitate and incubated the product at 4 °C for 10 min with rotation. The samples were sheared using the Covaris protocol (Peak power-75, Duty Factor-10, Cycles/Burst-100) for 10 min, and the lysate was immediately spun at a maximum speed at 4 °C for 10 min to collect the supernatant. We added an equal volume of NP-40 buffer to the supernatant, which contained 0.5 mmol/l DTT, 1 × PIC (Roche, 4693132001), and 100 U/ml RNaseOUT (Thermo Scientific, Massachusetts, USA 10777019). A 0.45 μmol/l syringe filter was used for the mixed solution and 50 μl of the lysate was removed as an input sample. Then, 25 μl of transfer protein G beads (MCE, HY-K0202) was added per 5 mol/l cells to each 1.5-ml tube, and they were placed on a magnet to remove the bead buffer. A total of 1 ml of the newly prepared NP-40 mixing buffer was added to wash the surface of the magnetic beads twice, then the sample was resuspended to 25 ul. Thereafter, we ‘precleared’ the filtered lysate by incubating with the above-mentioned protein G beads and rotating for 30 min at 4 °C. The sample was placed on the magnet, and the cleared lysate was transferred to a new 1.5-ml tube for each IP condition. After the lysate was thawed on ice and 5 μg of HuR antibody (HuR/ELAVL1 Rabbit mAb, A19622) was added to each sample, the lysate was rotated at 4 °C for 2 h. A total of 50 μl of protein G beads was processed according to the previous steps into the lysate and the beads were rotated at 4 °C for 1 h, before being placed on the magnet. The supernatant was removed, and 1 ml of the previously prepared NP-40 mixed solution buffer was added to wash the magnetic beads twice, rotating at 4 °C for 10 min each time. After the last wash, we removed the supernatant and froze the beads at −20 °C.
RNA purification and quantificationWe added 56 μl of RNase-free water and 33 μl of freshly prepared 3× reverse-crosslinking buffer, which consisted of 3× PBS (without Mg or Ca), 6% N-lauroyl sarcosine, 30-mmol/l EDTA, 15-mmol/l DTT, 10-μl proteinase K, and 1-μl RNaseOUT, in order to resuspend the beads, and reverse crosslinking was performed at 42 °C for 1 h, and then at 55 °C for 1 h. We followed the instructions of RNA Clean & Concentrator TM5 (cat. No: R1014) to clean 100 ul of RNA solution, and we finally added 15 μl of enzyme-free water directly to the column matrix before centrifugation.
RNA isolation and RT-qPCRThe sample could be completely lysed by Trizol reagent (Takara, Tokyo, Japan, T9108). Following the instructions, chloroform was added to the lysate, and then mixed and centrifuged to form a supernatant layer, an intermediate layer, and an organic layer. The supernatant layer was collected with the RNA distribution and precipitated with isopropanol to recover the total RNA extracted into the cells. A cytoplasmic and nuclear RNA purification kit (Norgen, Ontario, Canada, 21 000) was used to isolate and purify the cytoplasmic and nuclear RNA. According to the HiScript Q RT SuperMix for qPCR (Vazymy, Nanjing, China, R123) kit’s instructions, RNA (1 μg each) was used to remove genomic DNA and reverse transcribed into cDNA. In order to use cDNA for quantitative PCR, following the instructions, we used a ChamQ Universal SYBR qPCR Master Mix (Vazymy, Nanjing, China, Q711) to remove cDNA (1 μl) and diluted it 20 times with dd-H2O for PCR amplification. The reaction conditions were predenaturation at 95 °C for 30 s, and then cycling (denaturation at 94 °C for 10 s, annealing and extension at 60 °C for 30 s) 40 times. The experiment was repeated three times and each sample provided two replicate wells. The internal reference for COX-2mRNA and TUG1 was phosphoglycerate kinase (PGK). Moreover, 2−ΔΔct was used to analyze the data.
Primer sequence
TUG1 F: GAGACACGACTCACCAAGCA R: GAAGGTCATTGGCAGGTCCA
PGK F: TGCACGCTTCAAAAGCGCACG R: AAGTCCACCCTCATCACGACCC
COX-2 F: TTCAACACACTCTATCACTGGC R: AGAAGCGTTTGCGGTACTCAT
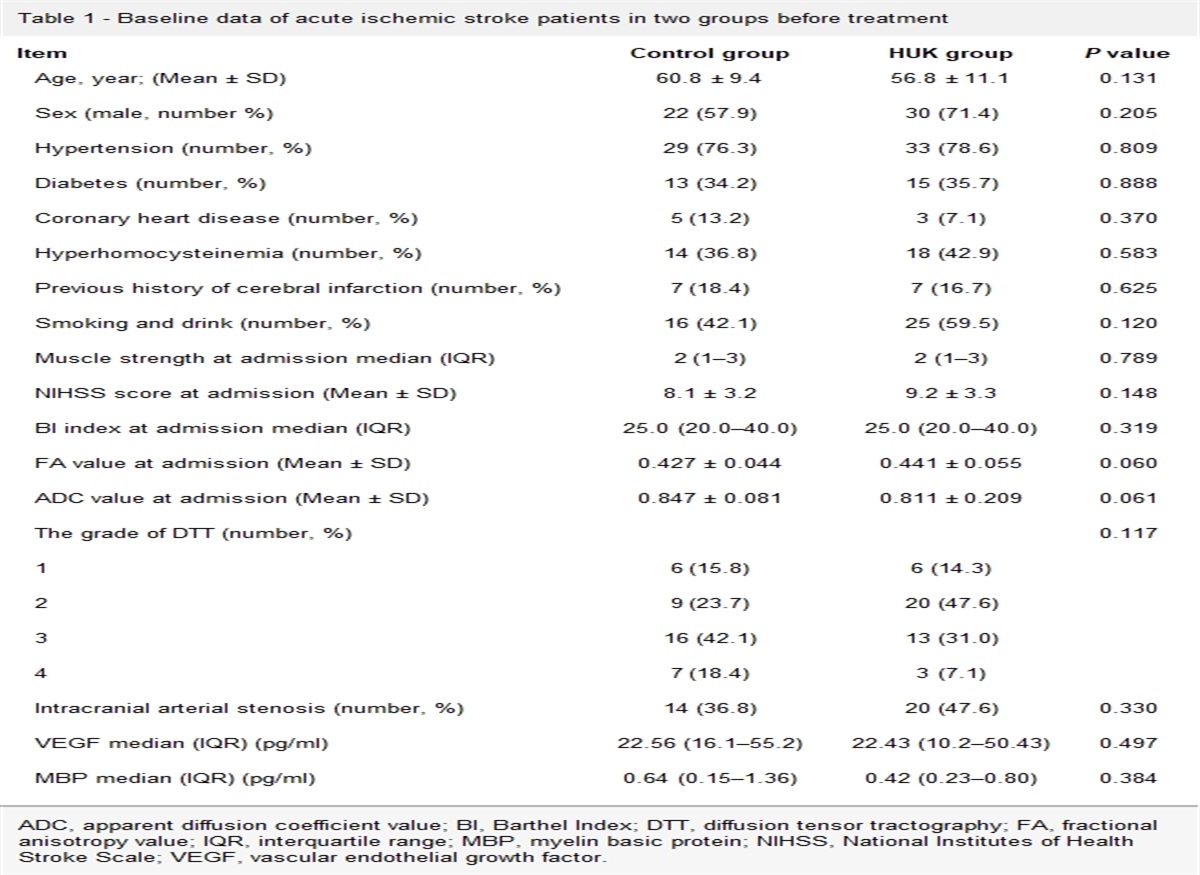
Bioinformatics analysisA bioinformatics analysis was performed to investigate potential HuR-binding sites in the lncRNAs sequences and three different in-silico approaches were used (Table 1). First, the RNA-protein interaction prediction (RPISeq, http://pridb.gdcb.iastate.edu/RPISeq/) was used to test the possible interaction between the TUG1 and HuR, with random forest (RF) values and support vector machine (SVM) values as the results for the likelihood of mutual binding. A score of more than 0.5 was considered ‘positive’ (following the guidelines provided by the website) for possible interaction.
Table 1 - The top 10 targets that potentially interact with TUG1 Protein ID Random forest (RF) Support vector machine (SVM) Protein region RNA region Interaction propensity Normalized score ELAVL2 (HuR) NP_001361165.1 0.85 0.96 218–269 162–445 94.4 6.08 Celf1 NP_001231820.1 0.7 0.98 26–77 175–458 101.36 5.67 Hnrnpc NP_001347110.1 0.7 0.88 201–252 175–458 115.01 5.94 Fmr1 NP_001361648.1 0.75 0.96 393–444 175–458 98.35 5.47 Srsf4 NP_001356237.1 0.8 0.97 271–322 175–458 85.88 5.61 Tlr3 NP_001344245.1 0.75 0.99 705–756 175–458 103.37 5.51 Pabpc1 NP_032800.2 0.7 0.99 236–287 175–458 100.13 5.46 Apc NP_001347909.1 0.7 1 1882–1997 175–458 98.98 5.10 Suz12 NP_001156490.1 0.65 0.99 567–627 175–458 101.02 5.48 Ptbp2 NP_001297640.1 0.75 0.99 56–107 175–458 91.87 5.03Second, the RBPmap database (http://rbpmap.technion.ac.il) was used to identify the number of potential interaction sites obtained with a ‘high stringency’ filter. The RBPmap database (only compared to human proteins, but protein sequences were conserved across species) was used to identify and score the obtained potential interaction sites. From this, we derived all the highest-scoring potential protein binding sites. Finally, the lncRNAs sequences were aligned to the RNAbp database (RBPDB, http://rbpdb.ccbr.utoronto.ca/index.php). This database allows for the identification of all potential RNA-binding sites, using a default threshold score of 0.8 (indicated by the website as the optimal cutoff in order to have a ‘confident’ score of the lncRNAs-protein interaction). All the RNAs analyzed showed confident HuR-binding sites, which were analogous to the U-rich sequences bound by HuR. Together, the three approaches demonstrated a real potential interaction between the TUG1 and HuR.
The CatRAPID fragments module is introduced to identify TUG1 regions involved in protein binding. The RNALFold algorithm from the Vienna package is employed to select RNA fragments in the range 100–200 nt with predicted stable secondary structure. Secondary structure stabilities are segments that have lower free energy for the higher number of bases that can be paired, the choice of segments in the range of 100–200 nt is optimal because it allows simultaneously: (a) selection of secondary structures with comparable free energy and (b) high sequence coverage (>90%) for long transcripts such as TUG1. The interaction fragments algorithm is a variant of the RNA interaction strength algorithm that allows the identification of putative binding areas in long sequences.
CatRAPID graphic module enables a quick assessment of the interaction propensity of a protein-RNA pair. The interaction propensity measured the interaction probability between 1 protein (or region) and 1 RNA (or region). This measure is based on the observed tendency of the components of ribonucleoprotein complexes to exhibit specific properties of their physicochemical profiles that can be used to make a prediction.
The concept of interaction strength is introduced to compare the interaction propensity of a protein-RNA pair with a reference set that has little propensity to bind (random associations between polypeptide and nucleotide sequences). Reference sequences have the same lengths as the pair of interest to guarantee that the interaction strength does not depend on protein and RNA lengths. For each protein-RNA pair under investigation, we use a ‘reference set’ of 102 protein and 102 RNA molecules (a total of 104 nonredundant protein-RNA pairs), randomly associated between peptide and nucleotide sequences, to determine potential interaction strengths. CatRAPID strength module allows for evaluating the significance of the interaction of a protein-RNA pair by comparing the result with a reference set of 104 interactions. The graphical representation of the CDF distribution value indicates the significance of the interaction propensity.
Statistical analysisAll experiments were independently repeated at least three times and analyzed using the GraphPad Prism software, Santiago, USA. Statistical differences were calculated using a two-tailed Student’s t-test, and all values are expressed as the mean ± SD. When P < 0.05, we consider the difference to be statistically significant. Ns: not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
ResultsThe expression of TUG1 is highly induced in the nucleus by OGD but decreases in the cytoplasm.
In order to detect the TUG1 expression pattern during OGD, we isolated the total RNA of mouse HT22 cell line treated by OGD and then identified the expression level of TUG1 using real-time quantitative polymerase chain reaction (qRT-PCR). We found that the expression of TUG1 in the HT22 cell line was significantly decreased in vitro after 4, 6, and 12 h of OGD treatment as compared with the control group (Fig. 1a). The cell viability was also assessed using a cholecystokinin-octopeptide-8 (CCK-8) assay, and, similar to previous findings, it was observed that OGD treatment significantly inhibited cell growth at 12 h as compared with the control group, which indicates that TUG1 expression may be associated with cell viability during OGD (Fig. 1b). In order to analyze the changes in TUG1 in different subcellular localizations in OGD, we divided the HT22 cell line into nuclear and cytoplasmic fractions and examined the changes in the subcellular localization of TUG1 during OGD. The results showed that TUG1 expression is highly induced in the nucleus (Fig. 1c), but significantly decreased in the cytoplasm after 6 h of OGD (Fig. 1d), indicating that TUG1 may have different functions in the nucleus and cytoplasm, or TUG1 that may shuttle between the nucleus and cytoplasm, leading to the different TUG1 distribution.
 Fig. 1.:
Fig. 1.: The expression level of TUG1 in neurons treated with OGD. Levels of TUG1 were tested using RT-qPCR assay. Normalization was performed with PGK. (b) Cell viability was measured using CCK-8 solution after treating cells with OGD for 12 h. (c and d) TUG1 levels in the nucleus and cytoplasmic compartments of HT22 cells were determined using qRT-PCR. PGK was used as the cytoplasmic control, and U6 snoRNA was used as the nuclear control. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. CCK-8, cholecystokinin-octopeptide-8; OGD, oxygen-glucose deprivation; PGK, phosphoglycerate kinase; qRT-PCR:real-time quantitative Polymerase Chain Reaction; TUG1, Taurine upregulated gene 1.
Taurine upregulated gene 1 interacts with HuR protein in HT22 cellsProtein-RNA interactions are ubiquitous and are critical aspects of many cellular processes, such as splicing, polyadenylation, trafficking, stability, and translation. Moreover, they are implicated in pathologies including autoimmune, metabolic, neurological, and muscular diseases [11]. Long noncoding RNAs typically exert their functions by binding to one or more proteins, are key to many cellular processes, and their dysregulation has been implicated in various pathologies. Studies show that TUG1 interacts with TRAF5 in rat diabetic nephropathy, and TUG1 overexpression promotes the degradation of TRAF5 protein and affects podocyte apoptosis [12]. To determine which protein interacts with TUG1 during OGD, we first searched online to predict the RBP-RNA interactions using the Tartaglia lab tool.
The top 10 targets that potentially interact with TUG1 are shown in Table 1, with HuR returning the highest score. First, the TUG1 sequences were aligned to the RBPDB. This database allows for the identification of all potential RNAbp-binding sites, using a default threshold score of 0.85 (indicated by the website as the optimal cutoff in order to have a ‘confident’ score of the lncRNAs-protein interaction). Second, TUG1-protein interaction prediction (RPISeq was used to test the possible interaction between TUG1 and the protein. The RF and SVM values were obtained by analyzing the TUG1 sequence and the possible protein sequence, which could be used as to predict the possibility of mutual binding. A score of more than 0.5 was considered ‘positive’ (following the guidelines provided by the website) for a possible interaction, with all proteins indicating a possible interaction with TUG1. Finally, the RBPmap database was used to identify and score potential interacting sites obtained through the ‘high stringency’ filter. All potential protein binding sites with high scores were analyzed, and the scores were calculated according to the reliability. The aforementioned database only allows for comparisons with human proteins, but this is conserved across species.
The interaction fragments algorithm is a variant of the RNA interaction strength algorithm that allows identification of putative binding areas in long sequences. The interaction profile represents the interaction score (y-axis) of the protein along the TUG1 sequence (x-axis), giving information about the transcript regions that are most likely to be bound by the protein. (Fig. 2a). To identify potential interactions between TUG1 and the HuR receptor, we used the CatRAPID online algorithm, which can rapidly predict RNA-protein interactions and domains to evaluate the interaction tendency of TUG1 and the HuR receptor based on the secondary structure, hydrogen bonding, and molecular interatomic forces. The DP values above 50% indicate that the interaction is likely to take place. Interestingly, the CatRAPID revealed that there existed an interaction between TUG1 and the HuR receptor with a DP value of 100% (Fig. 2b). We eliminated the length dependence by introducing a ‘reference set’ composed of protein and RNA sequences that have exactly the same lengths under investigation. The interaction fragments algorithm was used to predict TUG1’s ability to interact with HuR. In our calculations, we used a reference set of 102 protein and 102 RNA molecules, randomly associated between polypeptide and nucleotide sequences, to identify potential interaction strength. In particular, we predicted HuR to have a strong propensity to bind to TUG1 (protein interaction strength = 96%) (Fig. 2c). After determining that HuR has the highest possibility of interacting with TUG1, we further verified this using an fRIP assay, followed by qPCR with the primers targeting TUG1. There were significantly more TUG1 bound to the HuR protein during OGD treatment, compared with the controls (Fig. 2d). These data suggest that the HuR protein can directly bind to TUG1 and TUG1 exerts its function during OGD through interacting with HuR.
 Fig. 2.:
Fig. 2.: In the OGD condition, TUG1 bound to the RNA-binding protein HuR. Prediction of the interaction between TUG1 RNA and HuR. (a) The shades of red on the heatmap indicate the interaction score for the individual nucleotide. (b) The actual prediction results for TUG1 and HuR are shown on the heatmap. The x- and y-axes represent the indexes of the RNA and protein sequences, respectively. The colors of the heatmap indicate the interaction score (ranging from −3 to +3) of the individual amino acid and nucleotide pairs. The total sum represents the overall interaction score. The catRAPID identified the interaction between TUG1 and HuR with confidence (interaction propensity = 144 and discriminative power = 100%). (c) The interaction fragments algorithm was used to predict TUG1 ability to interact with HuR. Reference sequences have the same lengths as the pair of interest to guarantee that the interaction strength is independent of protein and RNA lengths. The interaction strength ranges from 0 (noninteracting) to 100% (interacting). Interaction strengths >50% indicate a propensity to bind. (d) The fRIP experiment indicated that more TUG1 binds to HuR in the HT22 cell line in the OGD experimental group as compared with the control group. Levels of TUG1 were tested using RT-qPCR assay. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. fRIP, formaldehyde RNA immunoprecipitation; HuR, human antigen R; OGD, oxygen-glucose deprivation; TUG1, Taurine upregulated gene 1.
HuR protein undergoes relocation during oxygen-glucose deprivationAs the functional activity of HuR is regulated by dynamic subcellular localization, this underlies the contribution of HuR to many disease states. For example, under normal cellular physiology, HuR is primarily located in the nucleus, and when exposed to intrinsic and extrinsic stress, HuR can translocate from the nucleus to the cytoplasm, where it stabilizes and increases translation of target mRNAs [8]. Therefore, we examined the expression and subcellular localization of HuR after OGD in the mouse HT22 cell line in WB experiments. The results demonstrate that the expression of total HuR protein in HT22 cells treated with OGD for 6 h was not significantly different from that of the control group (Fig. 3a and b). When the nuclear and cytoplasmic portions of HT22 cells were separated after OGD treatment for 6 h, we observed that the concentration of HuR protein in the nucleus was downregulated to 62% of that observed in the control group (Fig. 3c and d), whereas the concentration of HuR protein in the cytoplasm was significantly elevated (Fig. 3e and f). These results indicate that translocation of HuR protein from the nucleus to the cytoplasm may occur in HT22 cells under the stimulation of ischemia and hypoxia.
 Fig. 3.:
Fig. 3.: Comparative analysis of HuR protein expression in HT22 cells, the nucleus, and the cytoplasm after OGD treatment. (a and b) The concentration of HuR in HT22 treated with OGD for 6 h was measured by Western blotting. Normalization was performed using β-actin. (c and d) OGD cells tended to have lower HuR in the nucleus as compared with the control. Normalization was performed using H3 (total histone H3). (e and f) OGD cells tended to have higher HuR in the cytoplasm as compared with the control. Normalization was performed using β-actin. Data are shown as mean ± SEM.*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. HuR, human antigen R; OGD, oxygen-glucose deprivation.
Knockdown taurine upregulated gene 1 in the nucleus but not cytoplasm reverses HuR localization changesIn order to verify if TUG1 can regulate HuR protein translocation during OGD, we utilized two different methods to knockdown TUG1, that is ASO, which more effectively target nuclear TUG1 and downregulate expression levels in HT22 (Fig. 4a); and small interfering RNA (siRNA), which target TUG1 within the cytoplasm and silence TUG1 (Fig. 4b). Thus, the HT22 cells were divided into four groups for specific nuclear knockdown of TUG1: NC+con; NC+OGD; ASO-TUG1+OGD; and ASO-TUG1+con; and four groups for knockdown in the cytoplasm: NC+con; NC+OGD; si-TUG1+OGD; and si-TUG1+con. OGD can significantly reduce the accumulation of HuR in the nucleus; however, TUG1 ASO can reverse this effect and prevent the decrease in HuR protein levels in OGD cells (Fig. 4c and d). Correspondingly, OGD can significantly elevate HuR protein levels in the cytoplasm, and knockdown TUG1 in the nucleus can reduce the accumulation of HuR in the cytoplasm during OGD (Fig. 4e and f). Moreover, decreasing TUG1 in the cytoplasm during OGD using siRNA was not able to reverse the reduction in HuR in the nucleus (Fig. 4g and h) or the increase in HuR in the cytoplasm (Fig. 4i and j). All these results shed light on the mechanisms through, which nuclear TUG1 facilitates the shuttle of HuR protein from the nucleus to the cytoplasm under OGD experimental conditions.
 Fig. 4.:
Fig. 4.: Downregulation of TUG1 in the nucleus using ASO prevents the shuttling of the HuR protein from the nucleus to the cytoplasm, but not for TUG1 siRNA. (a and b) RT-PCR analysis showing that the level of TUG1 in the OGD+ASO-TUG1 group was significantly lower than in the ASO-TUG1 group, and that there was no significant difference between the OGD+si-TUG1 group and the si-TUG1 group. (c and d) The WB analysis showed that the HuR protein nuclear level was significantly decreased in the OGD+NC group, and that the OGD+ASO-TUG1 was restored to the control level. (e and f) The WB analysis showed that the HuR protein level was significantly decreased in the OGD + NC group, and that the OGD + ASO-TUG1 recovered to the control level. (g and h) The WB analysis showed that the HuR protein nuclear level in the OGD + NC group was significantly decreased and that the OGD + si-TUG1 could not be restored to the control level. (i and j) The WB analysis showed that the HuR protein level was significantly decreased in the OGD + NC group, and that the OGD + si-TUG1 did not recover to the control level. Data are shown as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. ASO, antisense oligonucleotides; HuR, human antigen R; TUG1, Taurine upregulated gene 1.
Knockdown of taurine upregulated gene 1 in the nucleus significantly reduces the proportion of apoptosisIn order to explore the effects of TUG1 knockdown in the nucleus and cytoplasm on cell survival and apoptosis, a CCK-8 assay was used for HT22 cells under OGD conditions with TUG1 ASO or siRNA, separately. We observed that ASO-TUG1 was able to significantly reduce the proportion of apoptotic cells after 6 h-OGD, whereas si-TUG1 had no significant effect on the proportion of apoptotic cells under the same conditions (Fig. 5a and b). HuR shuttles to the cytoplasm and stabilizes its targets in response to various stimuli. COX-2 is a proapoptosis gene and potential HuR target gene, which possesses a long 3′-UTR containing multiple AREs that can be stabilized by HuR in various cell types [8]. In this study, fRIP-qPCR was used to further confirm that HuR can directly interact with COX-2 mRNA. The qRT-PCR results indicated a significant increase in COX-2 mRNA bound to HuR protein under OGD treatment, compared with the control group (Fig. 5c). The expression of COX-2 mRNA in the HT22 cell line was significantly increased in vitro after 4, 6, and 12 h of OGD treatment (Fig. 5d), which is associated with the decreased cell viability that we see in Fig. 1b. Since the HuR protein, shuttled by TUG1 from the nucleus to the cytoplasm, is responsible for the increase in COX-2 mRNA, the knockdown TUG1 from the ASO method should, therefore, also inhibit the increased expression of COX-2 in the cytoplasm. Indeed, we found that TUG1 ASO was able to partly reverse the increase in the COX-2 expression level (Fig. 5e). However, knockdown of cytoplasmic TUG1 with siTUG1 exhibited no significant change (Fig. 5f). These data suggest that LncRNA TUG1 may play a role in promoting neuronal apoptosis by facilitating the shift of the HuR protein from the nucleus to the cytoplasm.
 Fig. 5.:
Fig. 5.: Downregulation of TUG1 in the nucleus ameliorated HT22 cell apoptosis and reduced the mRNA expression of COX-2. (a) CCK-8 cell viability assays, performed to detect cell injury, showed that ASO-TUG1 promoted cell survival. (b) CCK-8 experiments showed that si-TUG1 had no significant effect on cell viability as compared to controls. (c) The fRIP experiment demonstrated a significant increase in COX-2 mRNA bound to HuR in the OGD experimental group. (d–f) Levels of COX-2 mRNA were tested using RT-qPCR assay. Normalization was performed with PGK. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. ASO, antisense oligonucleotides; CCK-8, cholecystokinin-octopeptide-8; HuR, human antigen R; OGD, oxygen-glucose deprivation; PGK, phosphoglycerate kinase; TUG1, Taurine upregulated gene 1.
DiscussionPrevious data showed that in the thromboembolic stroke model, infarctions most frequently take place in the cerebral cortex, hippocampus, and thalamostriate. People surviving episodes of cerebral ischemia often show a persistent memory deficit and cognitive decline. Therefore, investigating the pattern of ischemic injury in the hippocampus may provide insights into pathogenetic mechanisms and may help develop new therapeutic strategies. It is well known that the hippocampus is one of the brain regions most sensitive to ischemic damage and plays important roles in learning, memory, and epilepsy and is known to have a high susceptibility to ischemic damage compared with other brain structures in animals and humans. However, because hippocampal are most vulnerable to ischemia, cell death has been thought to represent a sensitivity of the neurons to injuries. To understand the role of hippocampal neurons in ischemia, we adopted a widely used cell model derived from immortalized parental HT4 cells to simulate the construction of a stroke model.
Many existing studies show that lncRNA can regulate cell processes and gene expression in cerebral IS injury. Thus, it could potentially play an important role in new treatment methods [5]. New technologies, such as RNA-seq, deep sequencing, and microarray analysis, have been used to screen out a large number of abnormally expressed lncRNAs in IS patients or animals with ischemic injury. These play an important role by regulating cell survival, inflammatory processes, and angiogenesis [13]. TUG1 polymorphism studies show that specific promoters can bind to the transcription factor globin transcription factor 1 (GATA-1) to increase TUG1 expression at the transcriptional level, and high TUG1 expression has been shown to indicate a similar risk in clinical studies to other known IS factors, including total cholesterol, triglycerides, HDL-cholesterol, and LDL-cholesterol [7]. TUG1 was reported to be upregulated in the caudate nucleus in Huntington’s disease and has recently been identified as a P53 target gene [14]. In the mouse hippocampal cell line OGD, wherein knocking down TUG1 increased the level of brain-derived neurotrophic factor (BDNF) and reduced apoptosis. Contrarily, TUG1 overexpression reversed the therapeutic effect associated with aerobic exercise [15]. In addition, in the HuR transgenic astrocyte mouse stroke model, the significant enhancement of angiogenic brain edema of astrocytes around the lesion represents a worse short-term functional node [16]. Therefore, the expression and activity of RBP may affect the treatment of IS, but whether it is regulated by lncRNA and participates in the pathophysiological mechanism of IS is unclear. Studies have shown that, in a mouse model of ulcerative colitis, TUG1 positively regulates the upregulation of c-myc by interacting with HuR to regulate cell apoptosis [17]. In this study, we propose that TUG1 may affect the inflammatory response in OGD through its interaction with HuR. Furthermore, we used the experimental method of fRIP to verify this hypothesis, showing that more TUG1 was bound to the HuR protein during the OGD process.
As compared with mRNA, lncRNA has a wide range of variation and a shorter half-life; in particular, nuclear-localized lncRNA is more unstable than other subcellular localized lncRNA [18], suggesting that lncRNA has a complex metabolism and diverse functions. Interestingly, we found a decrease in TUG1 mainly occurs in the cytoplasm of HT22 cells under ODG conditions. We then further tested the relative distribution level of HuR in the cytoplasm/nucleus after ischemia and hypoxia treatment. Our data show that both TUG1 and HuR distributions change significantly with OGD damage. Notably, under OGD conditions, TUG1 forms a complex with HuR and translocates from the nucleus to the cytoplasm. In addition, we confirmed that TUG1 knockdown in the nucleus with ASO decreased the apoptosis rate under hypoxic ischemia conditions. When TUG1 is inhibited in the nucleus, the transfer of HuR from the nucleus to the cytoplasm is significantly weakened, but there is no such phenomenon when TUG1 is inhibited in the cytoplasm. In addition, we confirmed that TUG1 is able to interact with HuR and carries it to the cytoplasm under OGD conditions, and then, HuR further increases the expression of COX-2 by binding to the UTR region of COX-2 mRNA.
HuR is a well-studied RNA-binding protein, which functions in the cytoplasm to promote the stability of ARE-mRNA. A number of studies have demonstrated that stroke is a comprehensive, multifactorial disease with multiple biological pathways. RNA-binding proteins are able to directly or indirectly affect cell apoptosis induced by ischemia and hypoxia. The RNA-binding protein QKI can inhibit cell apoptosis under the induction of myocardial ischemia by binding to the proapoptotic transcription factor FoxO1 and negatively regulating its downstream target genes [19]. In hypoxia-cultured cortical neurons and astrocytes, the expression of the TIAR protein (T-cell restricted intracellular antigen-related) was increased and colocalized with DNA damage in neuronal cells, suggesting that TIAR may be associated with brain ischemia [20]. MAARS has a preference for binding to HuR and assists in nucleocytoplasmic shuttling and regulates targeted apoptotic proteins, such as p53, p27, Caspase-9, and BCL2 in the cytoplasm, which is of great significance for atherosclerosis and a wide range of vascular disease states [10].
In our study, we found that COX-2mRNA increased significantly when HuR was enriched in the cytoplasm. However, after preventing HuR accumulation in the cytoplasm, COX-2mRNA expression declined significantly. In summary, our data indicate that knocking down TUG1 in the nucleus can inhibit HuR in the nucleus, weakening the stability of COX-2mRNA, which may further regulate AMPA glutamate receptors to inhibit inflammation. An increase in COX activity can lead to the increased release of prostaglandins and to ischemic neuron damage by free radicals. COX inhibitors, which are effective anti-inflammatory drugs, have been used after transient global cerebral ischemia in rodents to improve the delayed death of hippocampal CA1 neurons [21]. In addition, COX-2 gene expression-deficient mice exhibited a protective effect in terms of reducing brain damage in a middle cerebral artery occlusion experiment, indicating that inhibiting COX-2 may be an important factor in the reduction of glutamate neurotoxicity [22]. Indeed, previous studies reported COX-2 to be the upstream factor of glutamate excitotoxicity, affecting cell fate by regulating AMPA glutamate receptors [23].
We observed that COX-2–mediated apoptosis was not completely reversed when TUG1 in HT22 nucleus was knocked down under OGD conditions. On the one hand, many factors lead to apoptosis after ischemia and a cascade reaction occurs. Various complex factors contribute to apoptosis after IS and further aggravate brain injury, including oxidative stress, the toxicity of excitatory amino acids, excess calcium ions, and inflammation. Thus, the apoptosis observed in our research may not be solely mediated by COX-2. On the other hand, the transcription and translation of COX-2 could be regulated by factors other than HuR. Transactivators, including CREB, ATF, C/EBP, C-Jun, C-Fos, and USF are demonstrated to bind to the promoter region of COX-2 and regulate its expression in human fibroblasts and endothelial cells. p300 is essential for COX-2 transcriptional activation by proinflammatory mediators in fibroblast. Though the TUG1 ASO almost completely blocked the translocation of HuR from the nucleus to the cytoplasm and showed a significant effect on regulation of COX-2 expression level, ther
留言 (0)