
記住我
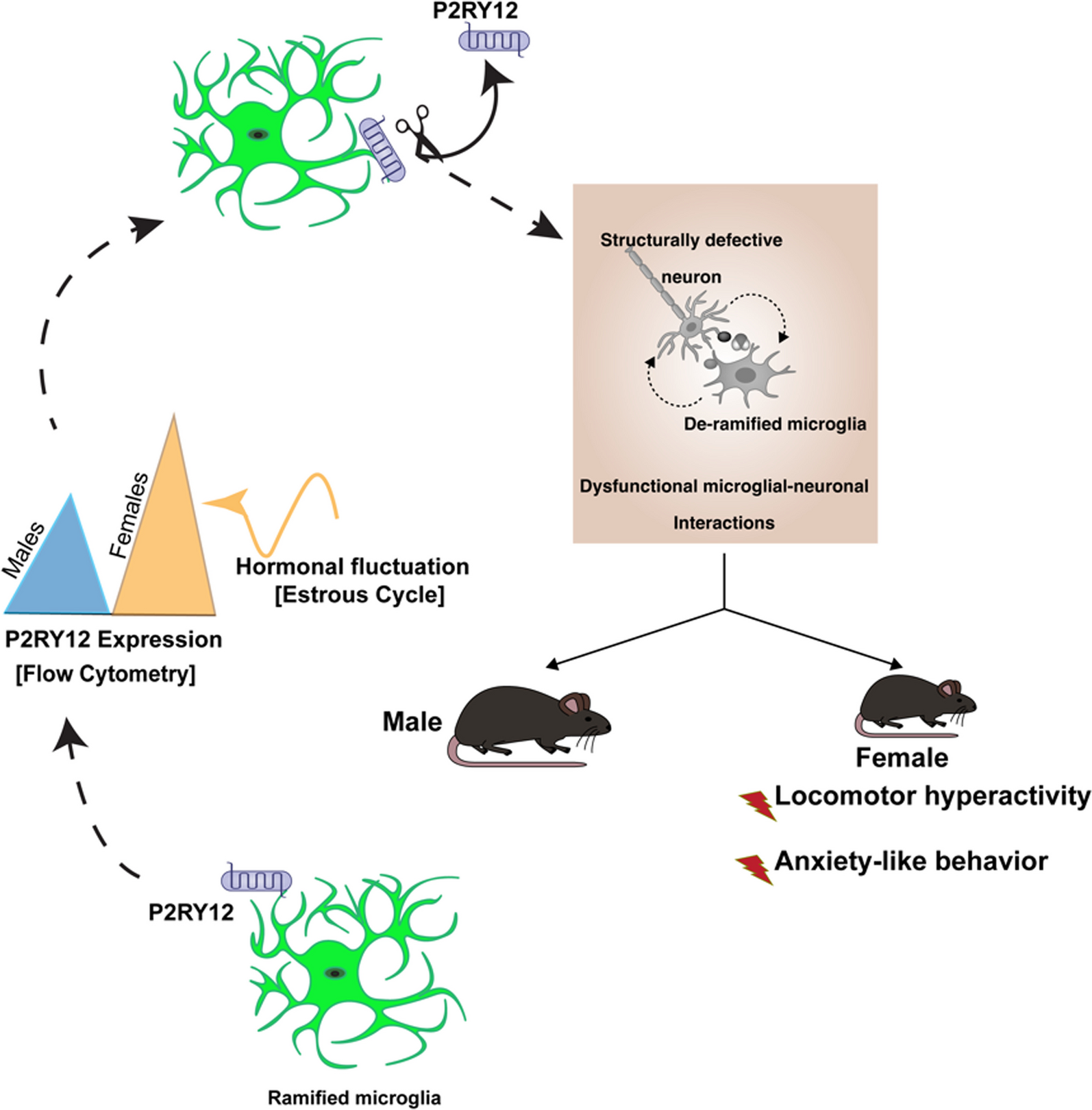

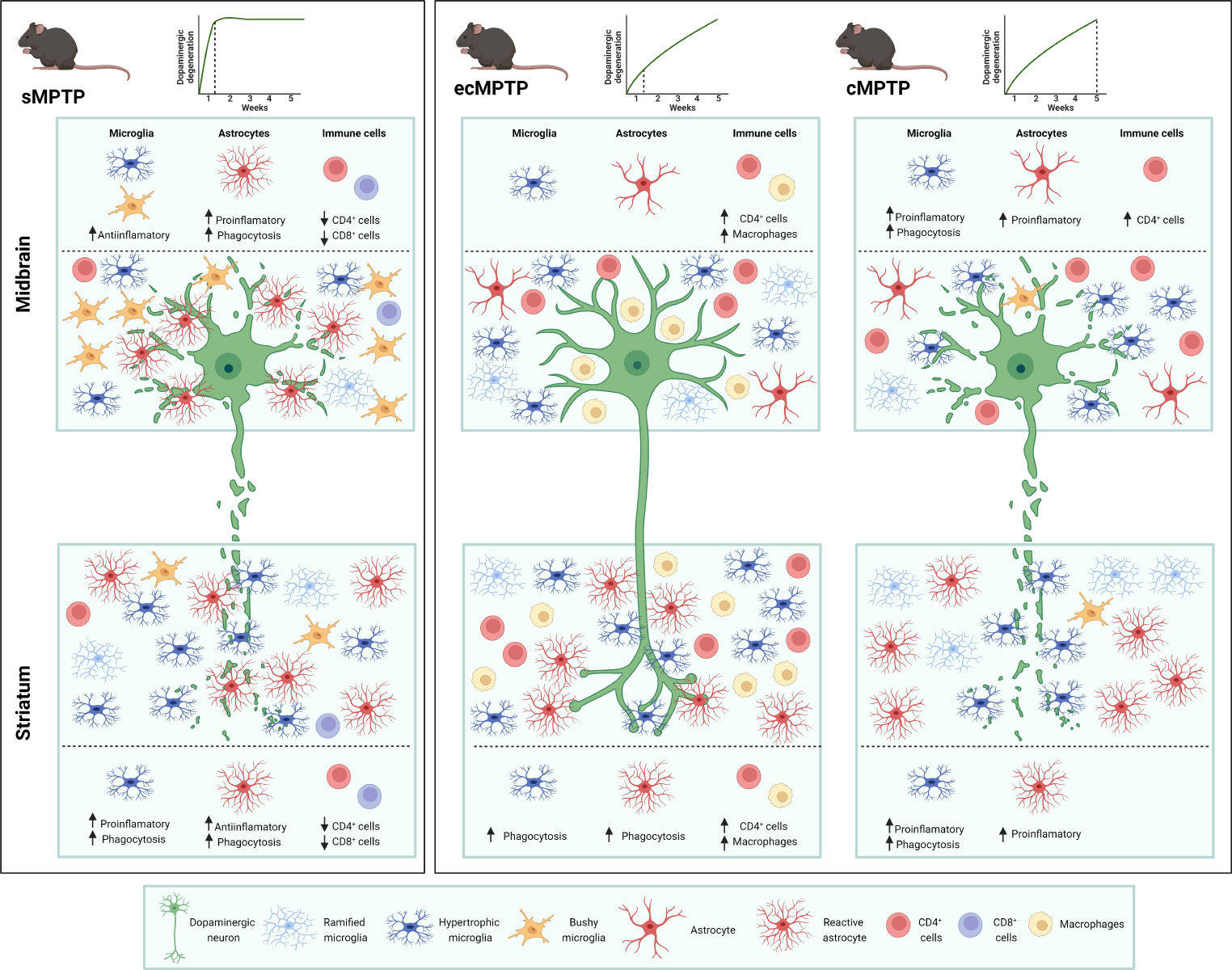

Generally, AR is hardly expressed in the normal tissue. However, there are upregulations of AR expression following stroke, non-alcoholic fatty liver disease, diabetic retinopathy as well as diabetic peripheral neuropathy [16, 19,20,21]. In our study, the expression of AR in the spinal cord of BPRA mice was compared with sham mice. AR was upregulated approximately 13-fold at the mRNA level and threefold at the protein level at 3 dpi (day post-injury) compared with the sham group (Fig. 1A–C). Immunofluorescence staining of spinal cord tissues also showed that the BPRA mice exhibit stronger AR expression than the sham mice at 3 dpi, and AR localization was predominantly cytoplasmic in neurons and microglia on the ipsilateral ventral horn of the spinal cord (Fig. 1D).
Fig. 1
BPRA mouse spinal cord showed increased AR expression. A, B Spinal cord AR protein expression normalized against a loading control (GAPDH) and C mRNA expression following BPRA injury. D Representative images showing elevated spinal AR levels indicated by immunofluorescence staining of neurons (NeuN), microglia (Iba1) and astrocytes (GFAP). The dashed lines in the ventral horn mark areas where MNs are situated. The white arrows indicate colocalization of AR (green color) with NeuN-, IBA1- or GFAP (red color)-positive cells. All data are presented as the mean ± SEM, n = 5 mice/group. *p < 0.01, **p < 0.01, ***p < 0.001, ****p < 0.0001 vs. sham, scale bar = 200 µm (a, e), scale bar = 50 µm (b–d, f–h)
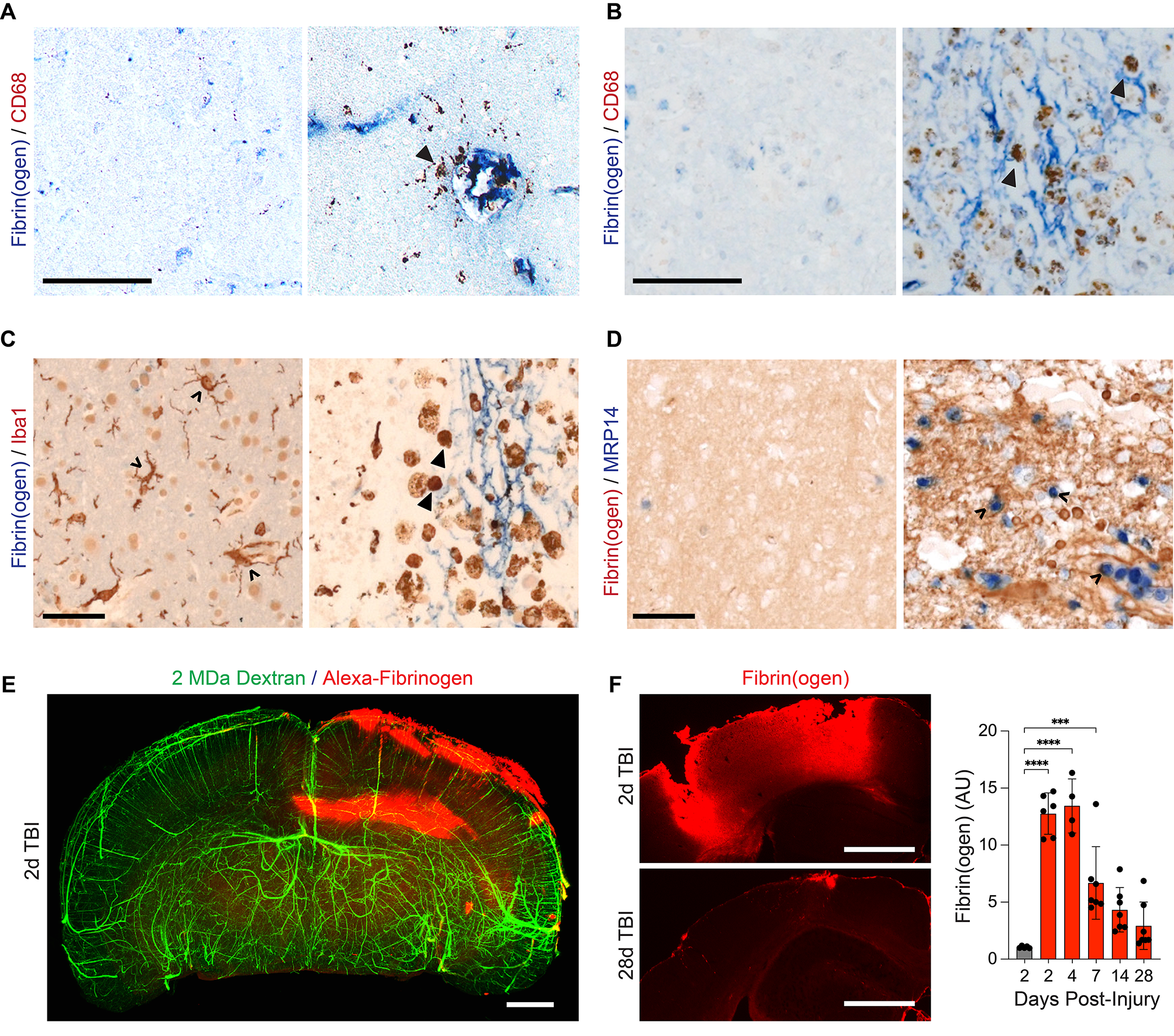
Genetic ablation of AR in mice prevented BPRA-mediated MNs deathTo investigate the effects of AR upregulation in BPRA, we sought to find out whether the deficiency of AR (examined using AR knockout mice, AR−/−) alleviated MNs death in the model of BPRA mice. Initially, we examined the effect of AR deficiency on MNs survival by staining neutral red, and the survival rate of injured MNs was assessed as the ratio of ipsilateral ventral horn neutral red-positive MNs to those on the contralateral ventral horn [22]. As shown in Fig. 2A and C, the survival ratio of MNs in AR−/− group was higher than WT group at 3, 7 and 14 dpi on the ipsilateral ventral horn of the spinal cord. Furthermore, AR−/− mice exhibited an increase in NeuN and anti-apoptosis gene Bcl-2 colocalization-positive neurons on the ipsilateral ventral horn of the spinal cord at 1–14 dpi (Fig. 2B, D). In addition, compared with the WT group, the AR−/− group showed more regeneration-related protein GAP43/NeuN colocalization-positive neurons at 7–14 dpi and higher GAP43 protein expression at 1–14 dpi on the ipsilateral ventral horn of the spinal cord (Fig. 2E, G, I, J). BPRA resulted in an increase in the apoptosis-related protein C-Caspase3/NeuN colocalization-positive neurons and C-Caspase3 protein expression level, which was attenuated in AR−/− mice at 1–14 dpi on the ipsilateral side of the spinal cord (Fig. 2F, H, I, K). Collectively, the results provided experimental evidence that genetic ablation of AR in mice prevented BPRA-mediated MNs death.
Fig. 2

Downregulation of apoptosis-related protein and upregulation of MNs survival rate in AR−/− mice after BPRA injury. A Spinal cords were counterstained with neutral red at 1–14 dpi to evaluate the MN survival rate. B Representative images showing Bcl-2 and NeuN staining at 1–14 dpi. C, D Summary of number changes in the ipsilateral spinal MN survival rate and Bcl-2 staining with quantification of NeuN-positive neurons between AR−/− and WT mice. E Representative images of MNs regeneration indicated by GAP43 staining with quantification of NeuN-positive neurons. F Representative images of MNs apoptosis in mice as measured by C-Caspase3 double staining with quantification of NeuN-positive neurons. G, H Summary of number changes in ipsilateral spinal C-Caspase3, GAP43 and NeuN colocalization-positive neurons between AR−/− and WT mice. I–K Representative images of spinal cord C-Caspase3 and GAP43 protein expression in AR−/− and WT mice normalized against a loading control (GAPDH). All data were analyzed by two-way ANOVA and are presented as the mean ± SEM, n = 5 mice/group, ####p < 0.0001, WT vs. sham group, ^^^^p < 0.0001, AR−/− vs. sham group, ****p < 0.0001, WT vs. sham group. Scale bar = 50 μm (A, B, E, F)
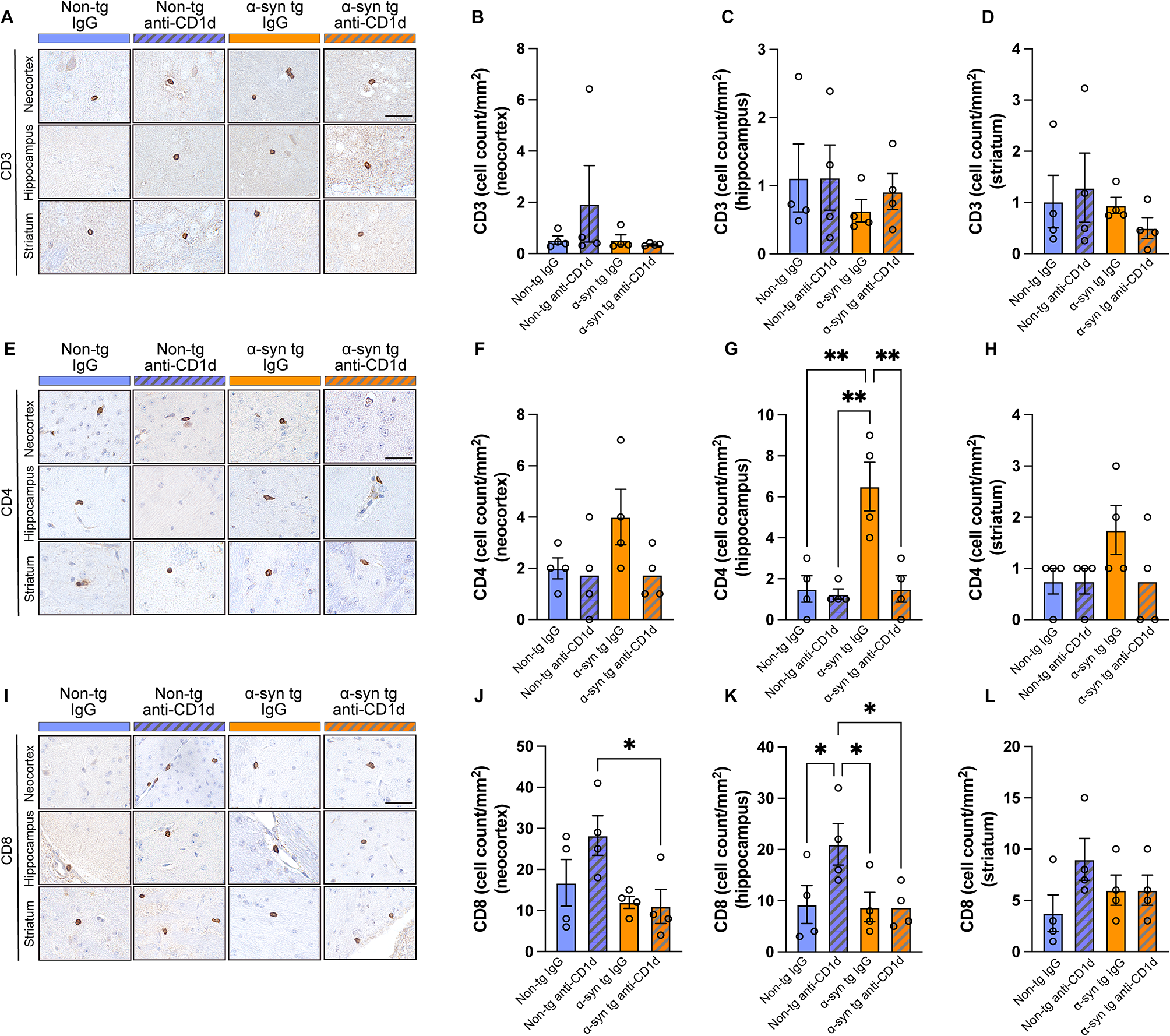
BPRA-mediated MNs death was attenuated by enhancing autophagy level in AR−/− miceThe dysregulation of autophagy is involved in motor neuron diseases, including spinal cord injury and amyotrophic lateral sclerosis. To evaluate the autophagy level after BPRA, we assessed the expression of the autophagy protein, LC3B in the injured spinal cord by immunofluorescence and western blotting. The conversion of LC3B-I to LC3B-II by adding phosphatidylethanolamine is important to autophagosome formation and this is considered as a marker of autophagosome formation and accumulation. Compared with BPRA WT mice, AR−/− mice had greatly increased LC3B/NeuN colocalization-positive neurons at 3–14 dpi on the ipsilateral ventral horn of the spinal cord and higher LC3B-II protein expression at 1–14 dpi on the ipsilateral side of the spinal cord (Fig. 3A, C, E, H), indicating the more accumulation of autophagosomes after injury in AR−/− mice.
Fig. 3
Protection against BPRA-mediated motor injury in AR−/− mice occurs by enhancing autophagy. A, B Representative images of autophagy hallmarks in mice as measured by LC3B and P62 double staining with NeuN. C, D Number changes in ipsilateral spinal LC3B/NeuN colocalization-positive neurons and P62/NeuN colocalization-positive neurons between AR−/− and WT mice. E–H Representative images of spinal cord Beclin1, P62 and LC3B protein expression normalized against a loading control (GAPDH) between AR−/− and WT mice. Data were analyzed by two-way ANOVA and are presented as the mean ± SEM, n = 5 mice/group, ####p < 0.0001, WT vs. sham group, ^^^^p < 0.0001, AR−/− vs. sham group, ****p < 0.0001, WT vs. sham group. Scale bar = 50 μm (B) and 20 μm (A)
To elucidate the autophagy mechanisms following BPRA, we assessed the levels of proteins that would regulate and form autophagosomes. Increased accumulation of LC3-II is likely due to either increased formation or decreased degradation of autophagosomes. Ubiquitinated cargo is delivered to autophagosomes by the adapter protein P62 (SQSTM1). During this delivery, the adapter protein (p62) is also degraded by autophagy alongside its cargo. Therefore, the accumulation of p62 suggests that autophagic degradation has been disrupted. Compared with BPRA WT mice, AR−/− mice had greatly decreased P62/NeuN colocalization-positive neurons at 3–14 dpi on the ipsilateral ventral horn of the spinal cord and lower P62 protein expression at 1–14 dpi on the ipsilateral side of the spinal cord (Fig. 3B, D, E, G). Moreover, western blot analysis showed more expression of the autophagy regulatory protein Beclin1 in AR−/− mice on the ipsilateral side of the spinal cord at 7 and 14 dpi than in WT mice, suggesting that the genetic ablation of AR increased initiation of autophagy (Fig. 3E, F). Overall, these results suggested BPRA-mediated MNs death was suppressed in AR−/− mice, which may be associated with enhanced autophagy level.
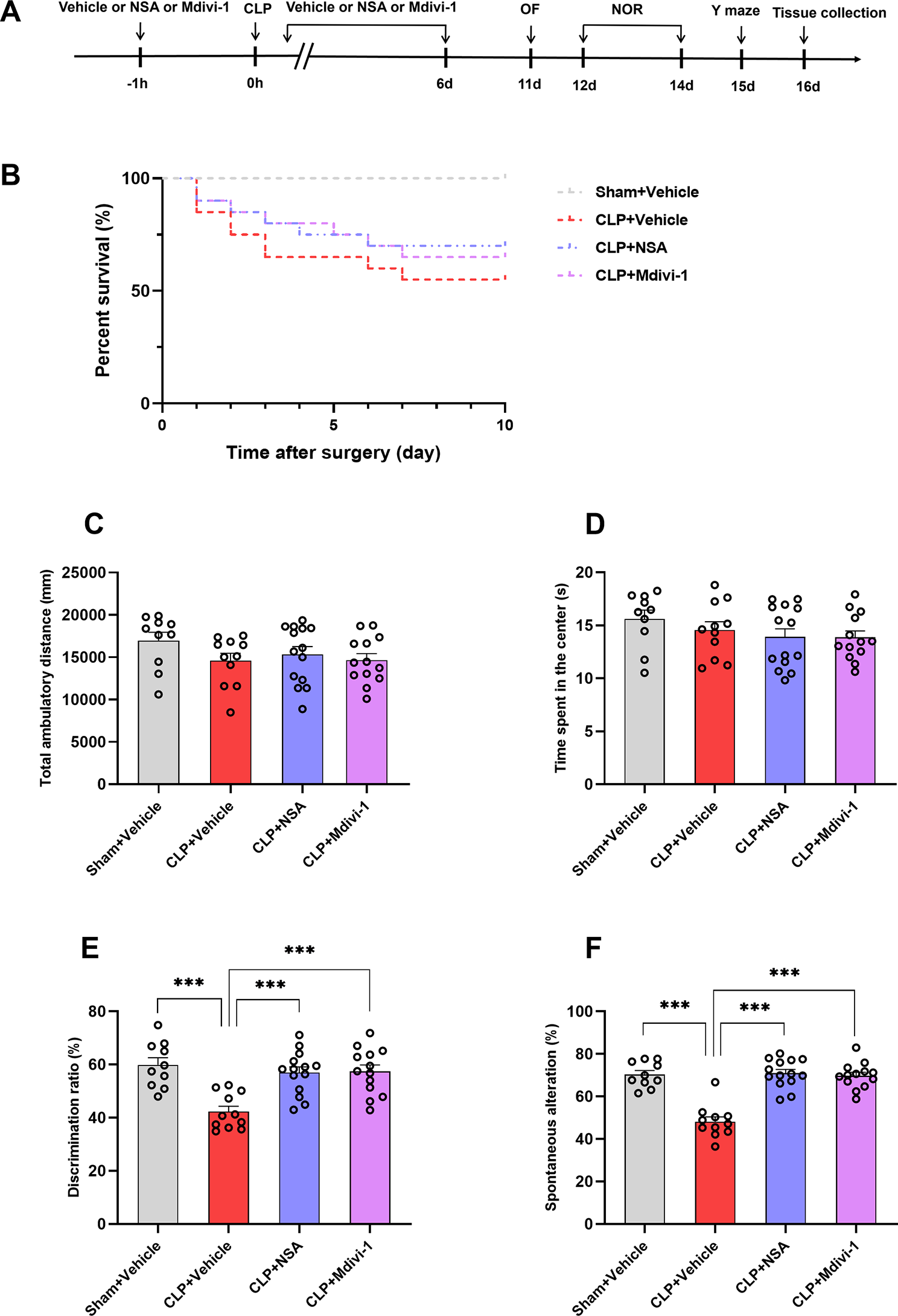
Autophagic vacuoles and mitochondria number were improved in a BPRA model of AR−/− miceWe further used electron microscopy (EM) to evaluate autophagic vacuoles number of MNs in both WT and AR−/− mice following BPRA. The structures of typical autophagic vacuoles are shown under high-magnification (13,500 ×) EM (green arrow in Fig. 4A). Compared with WT mice, the number of autophagic vacuoles was remarkably increased at 1–14 dpi in AR−/− mice (Fig. 4E). These results provided definite evidence that autophagy initiation was induced in AR−/− mice following BPRA injury.
Fig. 4
MNs display altered mitochondrial structure between AR−/− and WT mice at 1–14 d after BPRA injury. A Electron microscopy images of the ultrastructure of MNs in AR−/− and WT mice at 1–14 d after BPRA injury. Colored triangle indicate mitochondria, green arrows indicate autophagic vacuoles. Representative images of mitochondrial classes I (purple triangle), II (green triangle), III (blue triangle), and IV (orange triangle) (B) and their quantitative distributions (C) in WT and AR−/− mice. Quantitative graphs of the means ± SEM of mitochondria number/10 µm2 (D) and autophagic vacuoles number (E). Quantification was performed in at least 20 different fields (> 150 mitochondria) per mouse, n = 5 mice/group. ####p < 0.0001, WT vs. sham group, ^^^^p < 0.0001, AR−/− vs. sham group, ****p < 0.0001, WT vs. sham group
Mitochondria accommodate most cell energetic demands by generating ATP. Moderate autophagy is responsible for degrading damaged mitochondria caused by BPRA injury. We analyzed mitochondrial structures and number in the BPRA model at 1–14 dpi in WT and AR−/− mice. We first examined mitochondrial morphology and size by transmission electron microscopy and observed that there were larger mitochondria with altered crista organization in MNs from WT mice when comparing with those in AR−/− mice following BPRA (colored arrowheads in Fig. 4A). We further analyzed mitochondrial alterations in depth and classified mitochondrial morphology into four categories namely [23]: class I: fairly dark mitochondria, with a uniform matrix filled with densely packed and regularly distributed cristae; class II: mitochondria with disrupted cristae and a loss of matrix density; class III: empty mitochondria with disorganized cristae or cristae on the periphery; and class IV: swollen mitochondria with disrupted membranes) (Fig. 4B). Quantifications of mitochondrial subclasses were then performed, revealing that the WT mice at 14 dpi exhibited 4.2% mitochondrial class I, 4.9% class II, 10.4% class III and 80.5% class IV (Fig. 4C). However, the AR−/− mice at 14 dpi displayed a drastic increase in “healthy” mitochondria class I (78.5%) and class II (12.4%), class III (6.1%), class IV (3%) (Fig. 4C). We also observed that AR−/− mice also displayed a significant increase in the mitochondrial number at 1–14 dpi (Fig. 4D) compared with the WT mice following BPRA injury. Taken together, these findings demonstrated that genetic ablation of AR could eliminate damaged mitochondria and enhance autophagy level in BPRA mice mode.
Genetic ablation of AR in mice prevented the BPRA-mediated decrease in SIRT1–AMPK–mTOR signalingElevated AR activity is known to deplete cellular NADPH and cause high cytosolic NADH/NAD+ ratio. This results to loss of NAD+-dependent deacetylase SIRT1 activity [12]. Compared with WT mice, AR−/− mice had greatly increased SIRT1/NeuN colocalization-positive neurons at 3–14 dpi on the ipsilateral ventral horn of the spinal cord and higher SIRT1 protein expression at 1–14 dpi on the ipsilateral side of the spinal cord (Fig. 5A, C, E, F). Our results indicate that BPRA decreased the expression of SIRT1 and that AR−/− mice significantly restored the BPRA-induced decrease in SIRT1 expression.
Fig. 5
AR−/− mice show enhanced autophagy level by activating SIRT1–AMPK–mTOR signaling. A, B Representative images of SIRT1 and p-AMPK IF staining with NeuN-positive neurons. C, D Number changes in ipsilateral spinal SIRT1, p-AMPK and NeuN colocalization-positive neurons between AR−/− and WT mice. E–H Representative images of spinal cord SIRT1, p-AMPK and p-mTOR protein expression normalized against a loading control (GAPDH) in AR−/− and WT mice. Data were analyzed by two-way ANOVA and are presented as the mean ± SEM, n = 5 mice/group. ####p < 0.0001, WT vs. sham group, ^^^^p < 0.0001, AR−/− vs. sham group, ****p < 0.0001, WT vs. sham group. Scale bar = 50 μm (A, B)
A rising number of studies suggest that aberrant mTOR signaling impacts many pathways, such as glucose metabolism, energy production, mitochondrial function, cell growth and autophagy. AMPK and mTOR have a reciprocal relationship mediated by SIRT1, and the substrates of mTORC1 suppress autophagy [24]. We further measured the effect of AR ablation on BPRA-induced expression of AMPK and mTOR in motor neurons. Compared with WT mice, AR−/− mice had greatly increased p-AMPK/NeuN colocalization-positive neurons on the ipsilateral ventral horn of the spinal cord and higher p-AMPK protein expression at 3–14 dpi on the ipsilateral side of the spinal cord (Fig. 5B, D, E, G). Similarly, the BPRA-induced mTOR phosphorylation increase was also prevented in AR−/− mice at 1–14 dpi on the ipsilateral side of the spinal cord (Fig. 5H). It may infer that genetic ablation of AR potentially enhance SIRT1–AMPK–mTOR signaling and autophagy level following BPRA injury.
Protection against BPRA injury via attenuation of neuroinflammation and promoting microglia to switch from a pro-inflammatory to anti-inflammatory phenotype in AR−/− miceFollowing BPRA injury, the immunoreactive expression of Iba1 and GFAP, as markers of microglia and astrocytes, respectively, was used to assess neuroinflammation. Compared with the WT mice, the AR−/− mice had dramatically decreased Iba1 and GFAP average fluorescence intensity at 3–14 dpi on the ipsilateral ventral horn of the spinal cord (Fig. 6A–D). This evidence indicates that genetic ablation of AR attenuated ventral horn neuroinflammation in the injured spinal segment. Activated microglia can either become pro-inflammatory or anti-inflammatory phenotypes [25]. Compared with the WT group, the AR−/− group showed a dramatically decreased proportions of pro-inflammatory (CD16/32+/Iba-1+) microglia at 3–14 dpi (Fig. 7A, C) and an increased proportions of anti-inflammatory (Arginase1+/Iba-1+) microglia at 1–14 dpi on the ipsilateral ventral horn of the spinal cord (Fig. 7B, D). We further determined whether the AR-deficient mice also showed aberrant expression of cytokines that are functionally more important. Compared with WT mice, AR−/− mice had significantly reduced pro-inflammatory cytokines IL-1β and IL-6 levels at 1–14 dpi and inflammatory responses protein ICAM1 levels at 3–14 dpi, while increasing anti-inflammatory cytokine IL-10 levels at 1–14 dpi (Fig. 7E) on the ipsilateral side of the injured spinal cord. Moreover, compared with WT mice, AR−/− mice had significantly reduced pro-inflammatory cytokines IL-1β mRNA levels at 1–14 dpi and iNOS mRNA levels at 1–14 dpi, while increasing anti-inflammatory cytokines IL-10 mRNA levels at 3–14 dpi and Arg-1 mRNA levels at 3–14 dpi (Fig. 7F) on the ipsilateral side of the injured spinal cord. These results further indicate that genetic ablation of AR promotes microglia to switch from a pro-inflammatory to anti-inflammatory phenotype following BPRA injury.
Fig. 6
Attenuation of BPRA injury-induced neuroinflammation in the injured spinal cord in AR−/− mice. A, B Representative images of Iba1 and GFAP IF staining with NeuN. C, D Summary of changes in ipsilateral spinal IBA1 and GFAP average fluorescence intensity between AR−/− and WT mice. Data were analyzed by two-way ANOVA and are presented as the mean ± SEM, n = 5 mice/group, ####p < 0.0001, WT vs. sham group, ^^^^p < 0.0001, AR−/− vs. sham group, ****p < 0.0001, WT vs. sham group. Scale bar = 50 μm (A, B)
Fig. 7
Protection against BPRA injury by switching microglia from a pro-inflammatory to anti-inflammatory phenotype in AR-/- mice. A, B Representative images of CD16/32 and Arginase 1 IF staining with quantification of Iba1-positive microglia. C, D Summary of number changes in ipsilateral spinal CD16/32+ and Arginase1+ cells colocalization-positive to microglia between AR−/− and WT mice. E Summary of changes in ipsilateral spinal IL-1β, IL-6, ICAM1, and IL-10 levels between AR−/− and WT mice. F Summary of changes in ipsilateral spinal IL-1β, IL-6, Arginase 1, and IL-10 mRNA levels between AR−/− and WT mice. Data were analyzed by two-way ANOVA and are presented as the mean ± SEM, n = 5 mice/group, ####p < 0.0001, WT vs. sham group, ^^^^p < 0.0001, AR−/− vs. sham group, ****p < 0.0001, WT vs. sham group. Scale bar = 50 μm (A, B)
Genetic ablation of AR acts via 4-HNE–p-CREB signaling to exert its anti-inflammatory effectsConsidering cyclic-AMP response binding protein (CREB) plays a key role in anti-inflammatory microglial phenotypes and AR can reduce 4-HNE into GS-DHN, a macrophage inflammatory mediator, we estimated whether genetic ablation of AR acts via 4-HNE–p-CREB signaling to exert its “phenotype switching” effect following BPRA injury [26, 27]. Therefore, we sought to find out the expression of 4-HNE–p-CREB signal axis following BPRA in WT and AR−/− mice. Compared with the WT mice, the AR−/− mice had dramatically increased number of 4-HNE+/Iba1+ microglia at 1–14 dpi and number of p-CREB+/Iba1+ microglia at 1–14 dpi on the ipsilateral ventral horn of the spinal cord after BPRA injury (Fig. 8A–D). These results suggest that genetic ablation of AR exerts its anti-inflammatory effects via the 4-HNE–p-CREB signaling pathway.
Fig. 8
Upregulation of 4-HNE-p-CREB in AR−/− mice after BPRA injury. A, B Representative images of 4-HNE and p-CREB IF staining with microglia. C, D Number change in ipsilateral 4-HNE+ and p-CREB+ cells colocalization-positive to microglia between AR−/− and WT mice. Data were analyzed by two-way ANOVA and are presented as the mean ± SEM, n = 5 mice/group, ####p < 0.0001, WT vs. sham group, ^^^^p < 0.0001, AR−/− vs. sham group, ****p < 0.0001, WT vs. sham group
Testing the efficacy of pharmacological inhibition of AR in attenuating BPRA-induced autophagy disruption, neuroinflammation and MNs death in miceEpalrestat (EPAL), a pharmacological AR inhibitor with protective effects against diabetic complications in mouse models and human clinical trials [16], was used to evaluate the neuroprotective potential of AR suppression following BPRA injury. Significantly reduced AR, C-Caspase3 expression and increased ChAT, GAP43 expression on the ipsilateral ventral horn of the spinal cord, as evaluated using IF, were observed with pharmacological inhibition of AR by epalrestat at 14 dpi following BPRA injury (Fig. 9A). Furthermore, the administration of epalrestat significantly reduced the expression of P62, but greatly upregulated LC3B, SIRT1, p-AMPK expression on the ipsilateral ventral horn of the spinal cord at 14 dpi following BPRA injury (Fig. 9B). In addition, BPRA mice significantly reduced the expression of Iba1, GFAP and CD16/32 while greatly upregulating Arginase 1 expression on the ipsilateral ventral horn of the spinal cord at 14 dpi after treatment with epalrestat (Fig. 9C). Therefore, these results were similar to those of genetic knockout, suggesting that with use of epalrestat, pharmacological suppression of AR showed significant neuroprotective of MNs effects by attenuating BPRA-induced autophagy disruption and neuroinflammation (Fig. 10).
Fig. 9
Testing the efficacy of pharmacological inhibition of AR in attenuating BPRA-induced MNs death in mice. A (a–d) Representative IF staining images of AR, ChAT, GAP43, and C-Caspase3. (e–h) Number changes in ipsilateral ChAT+ MNs and AR+/NeuN+, GAP43+/NeuN+ and C-Caspase3+/NeuN+ colocalization-positive neurons between the BPRA and BPRA + EPAL groups in WT mice. B (a–d) Representative images of SIRT1, p-AMPK, LC3B and P62 IF staining with NeuN. (e–h) Number changes in SIRT1+/NeuN+, p-AMPK+/NeuN+, LC3B+/NeuN+ and P62+/NeuN+ colocalization-positive neurons between the BPRA and BPRA + EPAL groups in WT mice. C (a–d) Representative images of Iba1 and GFAP IF staining with NeuN and CD16/32 and Arg1 IF staining with Iba1. (e–h) Changes in Iba1 and GFAP average fluorescence intensity and number changes CD16/32+ and Arg1+ cells colocalization to Iba-1+ cells between the BPRA and BPRA + EPAL groups in WT mice. All data were analyzed by two-way ANOVA and are presented as the mean ± SEM, n = 5 mice/group, ####p < 0.0001, BPRA vs. sham group, ^^^^p < 0.0001, BPRA + EPAL vs. sham + EPAL group, ****p < 0.0001, BPRA + EPAL vs. BPRA group. Scale bar = 50 μm
Fig. 10
Schematic representation of the mechanistic role of AR in BPRA injury
留言 (0)