
記住我
This UK multi-institutional RCT was conducted from March 1, 2015, to January 31, 2019, as part of a UK National Institute of Heath Research-funded programme grant (PGfAR: RP-PG-0612-20001) aimed at developing the evidence base for the management of chronic constipation in adults, which is currently lacking [32].
A stepped-wedge randomized trial design permitted observer-masked data comparisons between patients awaiting intervention with those who had undergone surgery. Contrary to most stepped-wedge trials individual patients were randomized rather than utilizing cluster sampling. This is, in effect, a modification of a standard parallel-group, waiting-list control design, but with several advantages. First, a stepped-wedge design is more efficient and thus improves recruitment feasibility (the bane of nearly all surgical trials). Simulation demonstrated that a parallel-group design required a much larger sample size than that proposed for the current study at the same power. Second, the trial design meant that there was only a one-in-three chance (rather than one-in-two chance for a parallel group) of waiting 6 months for surgery, which was more acceptable to patients. The study received national ethical approval (15/LO/0609) and all patients provided their written informed consent. The study was registered with the ISRCTN Registry (ISRCTN11747152 [https://doi.org/10.1186/ISRCTN11747152]).
Eligibility criteria were: (1) age 18–70 years; (2) self-report of problematic constipation; (3) symptom onset > 6 months prior to recruitment; (4) symptoms meeting the American Gastroenterological Association definition of constipation [33]; (5) refractory constipation after a minimum basic standard (lifestyle and dietary measures and ≥ 2 laxatives or prokinetics) tried with no resolution of symptoms and no time requirement; (6) ability to understand written and spoken English (due to questionnaire validity); (7) ability and willingness to give informed consent; (8) failure of non-surgical interventions (minimum of nurse-led behavioral therapy) [34]; (9) IRP as determined by clinical examination and defecography, using the following criteria: (a) recto-anal or recto-rectal intussusception ± other dynamic pelvic floor abnormalities (e.g. rectocele, enterocele, excessive perineal descent); (b) deemed to be obstructing and/or associated with protracted or incomplete contrast evacuation by expert review. [35].
Exclusion criteria were: (1) significant organic colonic disease (red flag symptoms, e.g. rectal bleeding not previously investigated), inflammatory bowel disease, megacolon or megarectum (if diagnosed beforehand), severe diverticulosis/stricture/birth defects deemed to contribute to symptoms; (2) major colorectal excisional surgery; (3) current overt pelvic organ (bladder, uterus, and/or external rectal) prolapse or disease requiring obvious surgical intervention other than LVMR; (4) previous rectopexy; (5) sacral nerve stimulator in situ; (6) rectal impaction (as defined by digital and abdominal examination); (7) significant neurological disease (e.g. Parkinson’s disease, spinal injury, multiple sclerosis, diabetic neuropathy); (8) significant connective tissue disease (e.g. scleroderma, systemic sclerosis, systemic lupus erythematosus [not hypermobility alone]); (9) significant medical comorbidities and activity of daily living impairment (Barthel index ≤ 11); (10) major active psychiatric diagnosis (e.g. schizophrenia, major depressive illness and mania); (11) chronic regular opioid use (at least once daily) deemed to be the cause of constipation based on temporal association of symptoms with onset of therapy; (12) pregnancy or intention to become pregnant during study period; (13) known severe intra-abdominal adhesions.
Final review by pelvic floor multidisciplinary decision team (as per National Health Service [NHS] England recommendation) [36] to confirm appropriateness for surgery was performed for all patients.
Randomization and maskingParticipants were randomized to three arms with different delays before surgery (Fig. 1). In group I, LVMR was performed at T0; in group II, at 12 weeks (T12); in group III, at 24 weeks (T24). In all arms, there was a period of 4 weeks post-eligibility screening to arrange the logistics of surgery and ensure that patients had returned to their normal life routine after various assessments. Randomization was stratified by center. The Pragmatic Clinical Trials Unit (PCTU) at Queen Mary University of London developed a validated online randomization system, which was accessed by suitably trained and delegated researchers at recruiting sites and followed the PCTU-approved standard operating procedure for the study.
Fig. 1
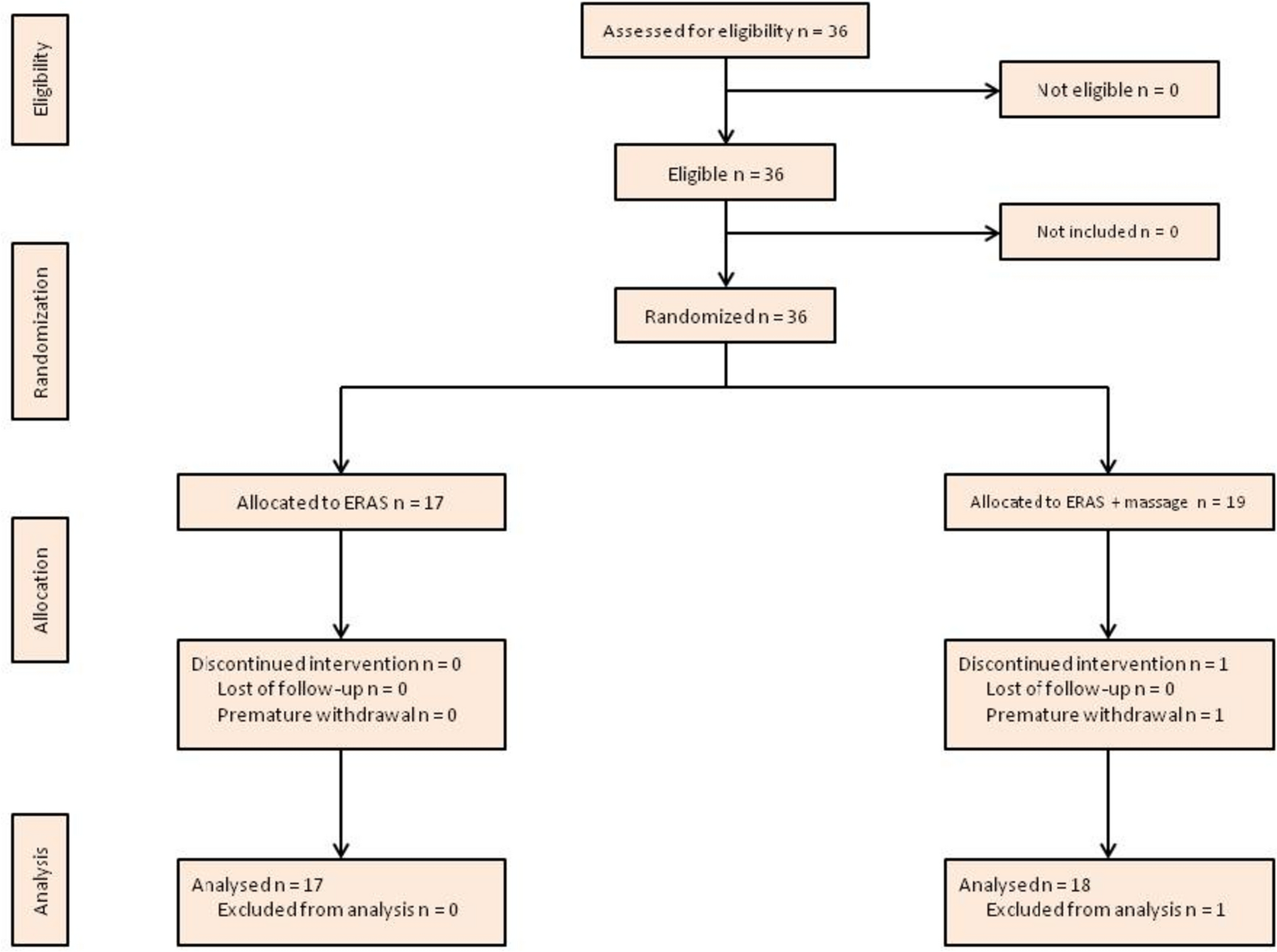
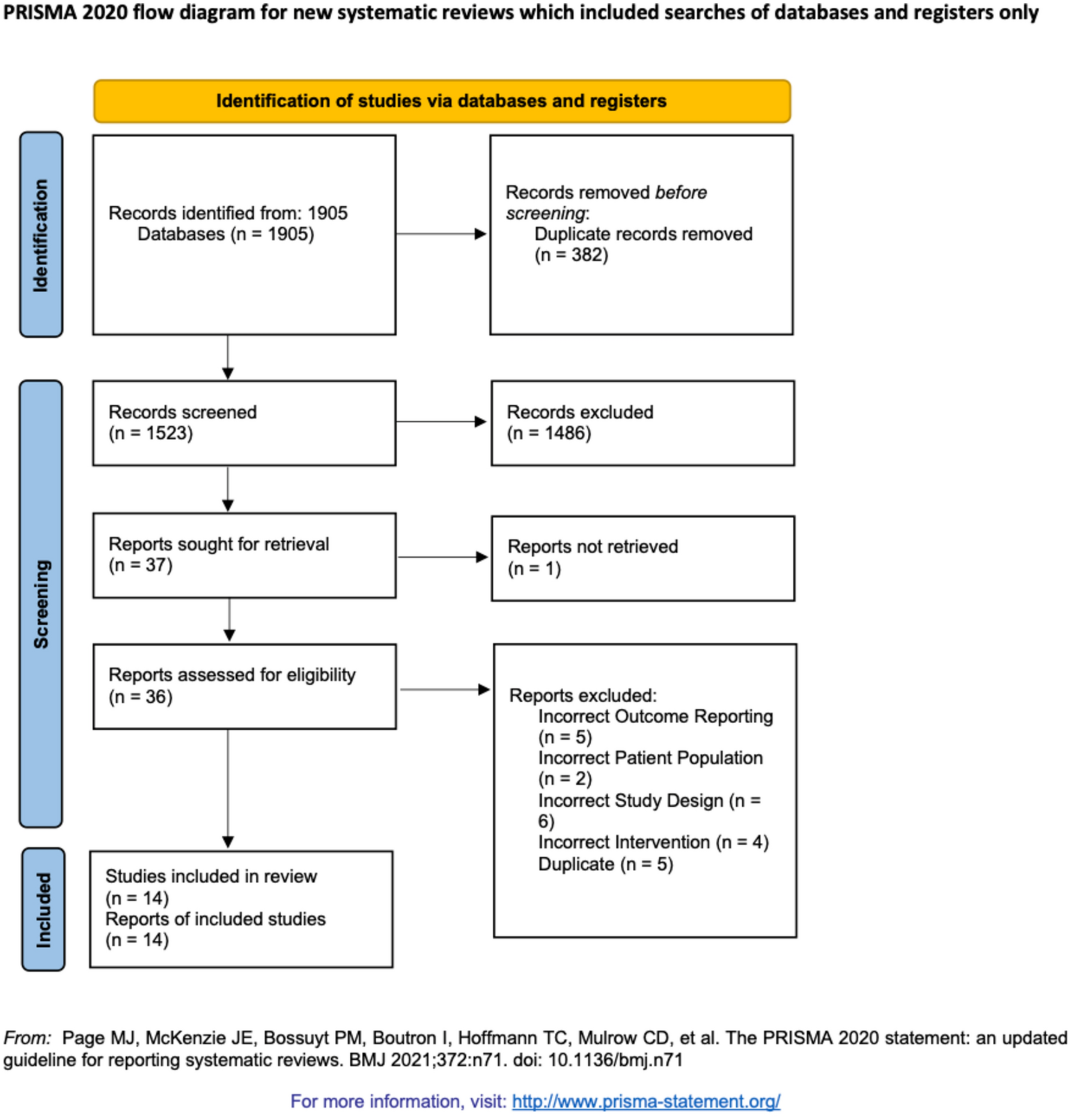
The CapaCiTY trial 3 Consolidated Standards of Reporting Trials (CONSORT) flow diagram. One patient did not undergo surgery; this patient continued to participate and was included in analysis on intention-to-treat principles
Patients and clinicians were necessarily aware of allocation to different waiting times. However, to minimize bias, a blinded researcher collected outcome data. For quantitative analysis, an analysis plan was developed and signed off by investigators and statisticians who were blind to allocation status and index intervention.
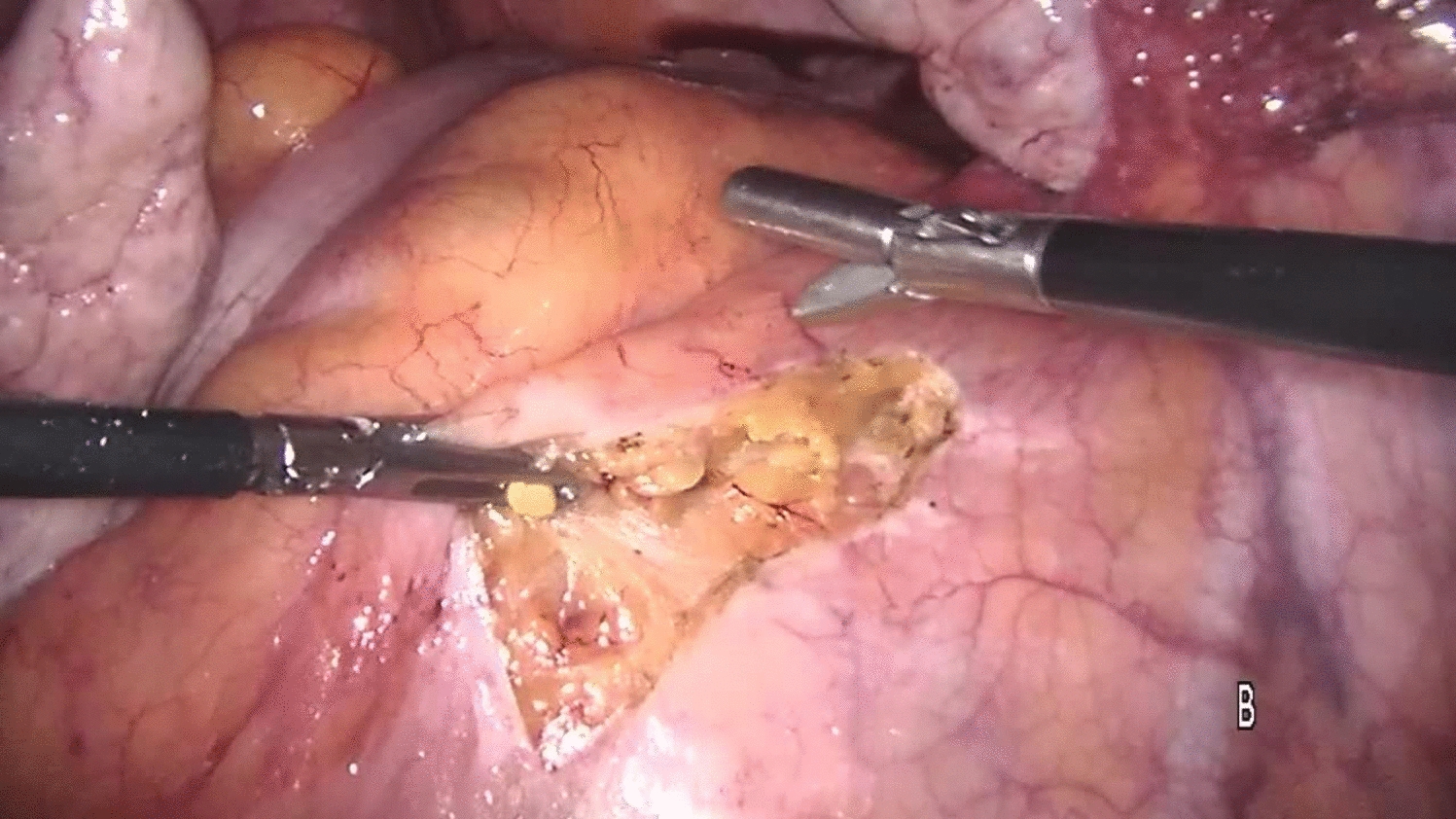
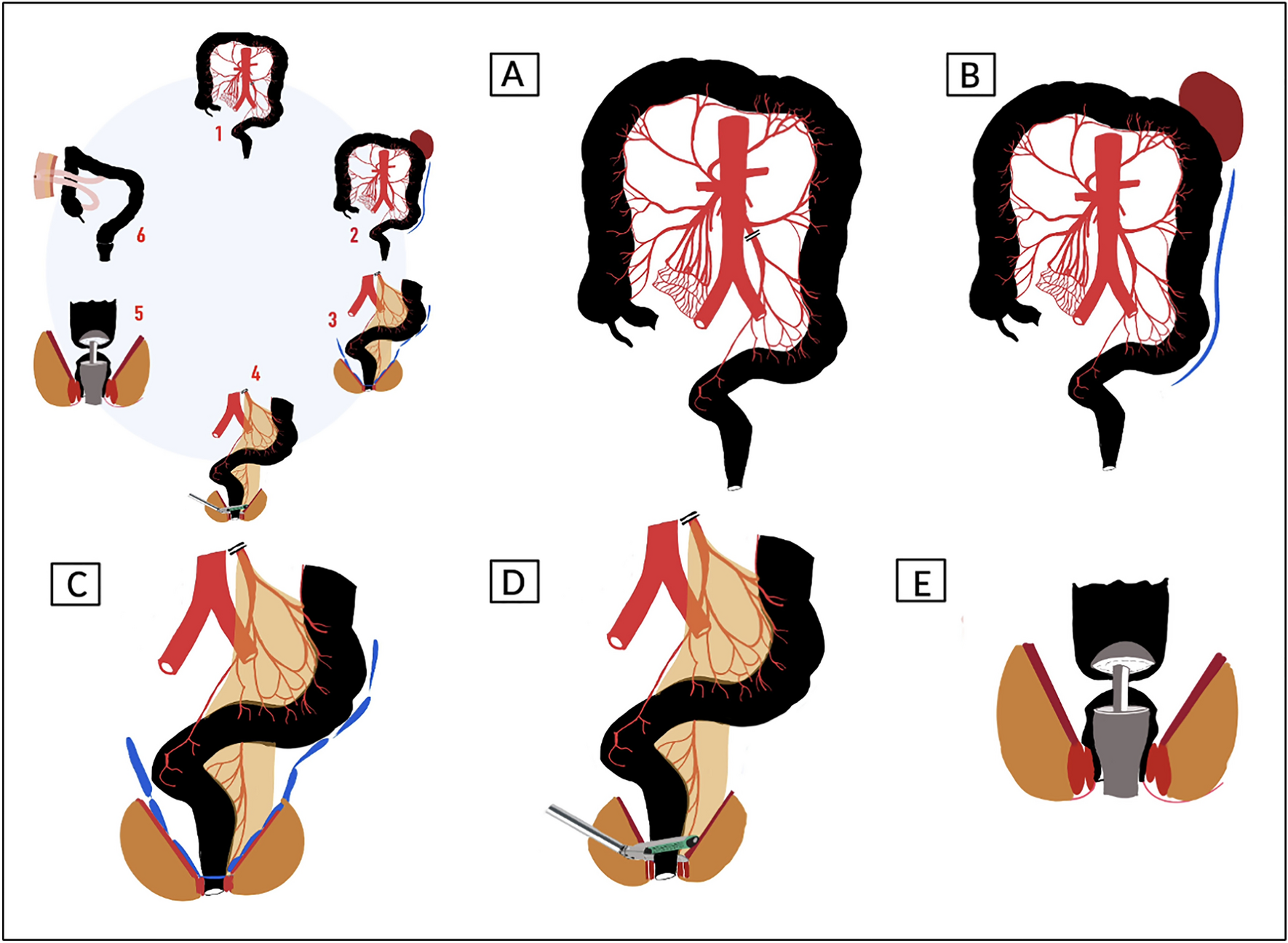
InterventionLVMR was performed according to a standard technique [31, 37, 38], starting with a peritoneal incision at the level of the sacral promontory and extending caudally (avoiding the hypogastric nerves along the side of the mesorectum) to the deepest part of the pouch of Douglas, and continued down the rectovaginal septum to the pelvic floor. The mesh was sutured to the ventral aspect of the distal rectum and further fixed to the lateral seromuscular borders of the rectum proximal and distal to the incised pouch of Douglas ± pelvic floor. If deemed necessary, the posterior vaginal fornix was elevated and sutured to the anterior aspect of the mesh to allow closure of the rectovaginal septum and correction of a mid-compartment prolapse (if present). The type of mesh inserted was left to surgeon’s choice (not being dependent on any specific clinical grounds). All participating surgeons had performed a minimum of 50 LVMR previously.
Surgery was performed as a day case or short stay procedure [39]. Postoperative management followed routine clinical care. Laxative use was standardized to a weaning course of macrogol transdermal delivery system (TDS) immediately postoperatively for 1 day, then reduced according to ease of bowel movements.
Surgical quality assessmentAdherence to the agreed procedural technique for the first included patient from each center was independently remotely assessed by a delegated surgical team provided by the Pelvic Floor Section of the Association of Coloproctology of Great Britain and Ireland. Monitoring and quality control were conducted remotely via video submission and assessed against the standardized LVMR protocol and defined assessment criteria [31, 37, 38]. Monitoring took the form of planned, random and triggered sessions (Supplementary Table 1).
OutcomesThe primary clinical outcome was Patient Assessment of Constipation Quality of Life (PAC-QOL) score [40]. This widely used, psychometrically robust measure of overall treatment response with concurrent validity to patient global ratings of success has been used by previous behavioral therapies and surgical trials, including LVMR [41], in chronic constipation [42]. For a chronic condition such as chronic constipation, a difference of 1.0 point in the primary outcome (score range = 1–4, with higher scores meaning higher negative effects on quality of life) was considered clinically important and also the minimum required to justify the cost and invasive nature of LVMR, or of a more complex and expensive treatment [43].
Secondary outcomes measures included 14-day diary data prior to each assessment (to record bowel frequency, whether each evacuation was ‘spontaneous’ [no use of laxatives] and/or ‘complete’, concurrent medication, health contacts, time away from normal activities including work, since the patient’s last visit), Generalized Anxiety Disorder scale (GAD-7) [44], Patient Health Questionnaire-9 (PHQ-9) [45], St Marks incontinence score [46], Pelvic Organ Prolapse/Urinary Incontinence Sexual Questionnaire (PISQ-12) [47], avoidant and ‘all or nothing’ behavior subscales of the chronic constipation Behavioral Response to Illness Questionnaire (CC-BRQ) [48], the Brief Illness Perception Questionnaire (BIPQ) [49], the EuroQol Visual Analogue Scale (EQ-VAS), the EuroQol Health Outcome Measure (EQ-5D-5L) [50], and the global patient satisfaction/improvement score on a five-point Likert scale. LVMR has a number of specific complications in addition to the general risks of surgery. These were recorded for outcome reporting. The study (not being of a medicinal product) did not record unrelated adverse events.
Participant, surgeon and research staff experience was investigated through individual digitally recorded telephone or in-clinic interviews up to 1 year after surgery with a purposively selected sample to represent a range of demographics. Separate consent was taken for interviews. Data were analyzed using a pragmatic thematic and qualitative analysis.
Follow-upThe study duration allowed for follow-up to a maximum of 96 weeks (i.e. 24 months) with data collection at 0, 12, 24, 36, 48 weeks post run in (stepped wedge) and thence at 12-week intervals within the cohort assessments at 60, 72, 84 and 96 weeks post run in. Thereafter, participants left the study and returned to ‘routine clinical care’ as determined within their local National Health Service institution (or were recruited to subsequent trials).
Statistical analysisThe sample size was calculated using the primary clinical outcome [40] by simulation using the ‘simsam’ package in Stata® V.14.2 (Stata Corporation, College Station, Texas, USA). Using a stepped wedge design, we hypothesized that PAC-QOL score at any time point during follow-up will be approximately 1.0 point lower (better) than preoperative participants. We assumed that PAC-QOL score followed a normal distribution over all time points with a standard deviation (SD) of 1.5 points and with a correlation between repeated assessments equal to 0.5 points. Simulation showed that detection of a 1-point difference in PAC-QOL score at 6 months with 95% power (purposely chosen to reflect the magnitude and risk of intervention) at the 5% significance level required 34 participants in each of the three arms. Allowing for a 10% loss to follow-up, a sample size of 38 was needed per arm, for a total sample size of 114 patients across the 3 arms. Should the correlation between repeated assessments be < 0.5 points, a sample size of 114 will still provide at least 90% power for the study. This was calculated using the same simulation procedure with correlations of 0.3 and 0.1 points.
The primary outcome was analyzed as a continuous variables on intent-to-treat basis at 24 weeks post-surgery. PAC-QOL scores at the time-points T0, T12, T24, T36, T48, T60, and T72 weeks post run in period in the three arms were analyzed using a mixed linear regression model, adjusting for a random effect of participant and a fixed effect of time since randomization, to estimate mean differences between PAC-QOL score before and after LVMR. To model the effects of surgery, dummy variables were used to indicate if participants had already received treatment before each follow-up time. The Kenward–Roger correction was employed to account for inflated type I error rates due to the small sample size. The contrast of primary interest was between the score at 24 weeks after surgery and the score at baseline. Some outcomes were scores calculated by summing the responses to all the questions in a questionnaire. If fewer than half of the questions were unanswered the missing responses were imputed with the mean of the available cases. All outcomes were analyzed under a ‘missing at random’ assumption (i.e. assuming that ‘missingness’ depended only on outcomes that had been observed). Patient Assessment of Constipation‐Symptoms (PAC-SYM) scores [51] were analyzed by the same approach as above. Binary outcomes were summarized at 24 and 48 weeks post-surgery with number and percent indicating problems and an odds ratio comparing 24 and 48 week outcomes to baseline. Data were analyzed using Stata® V.14.2 (Stata Corporation, College Station, Texas, USA).
留言 (0)