
記住我

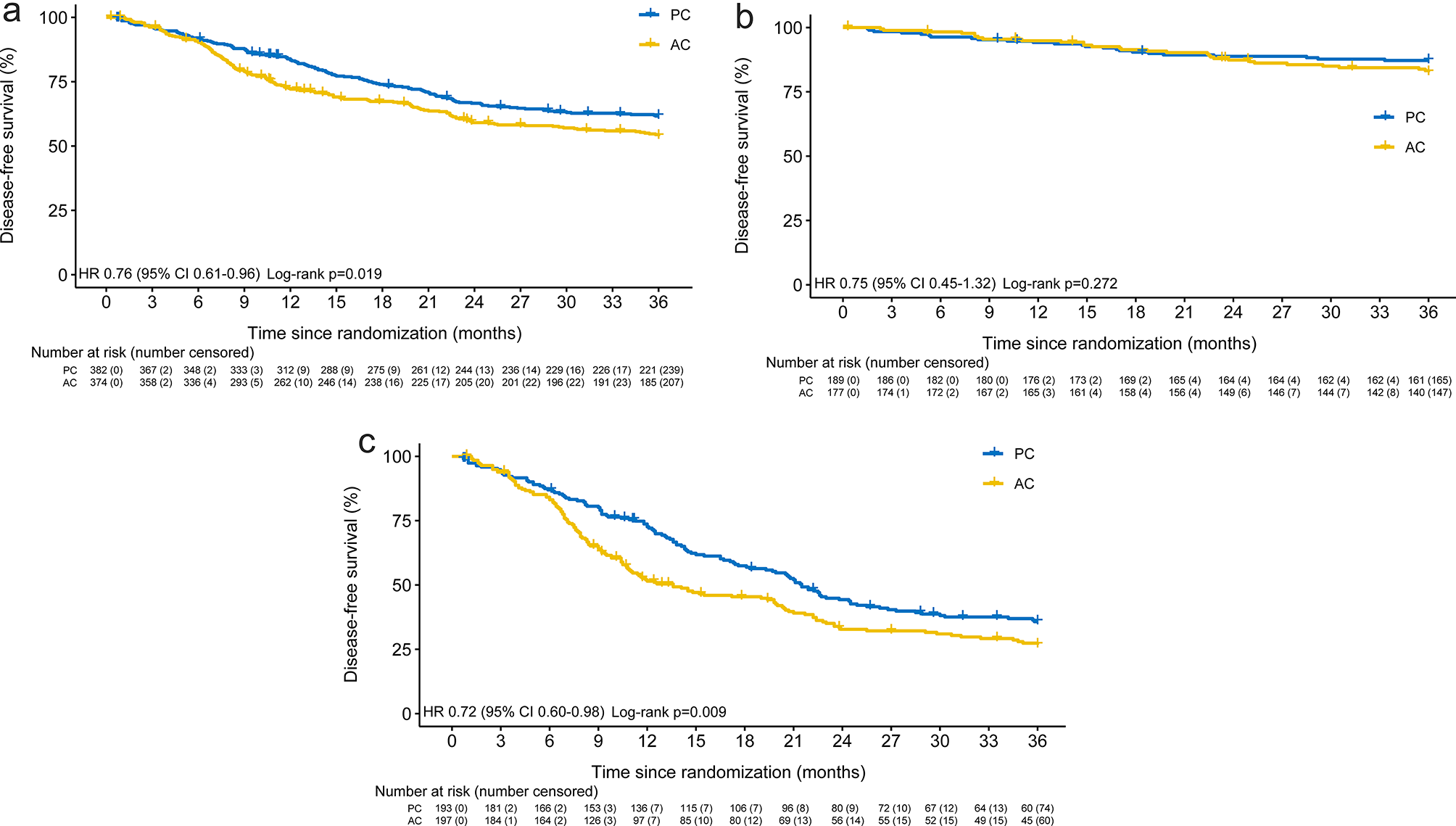
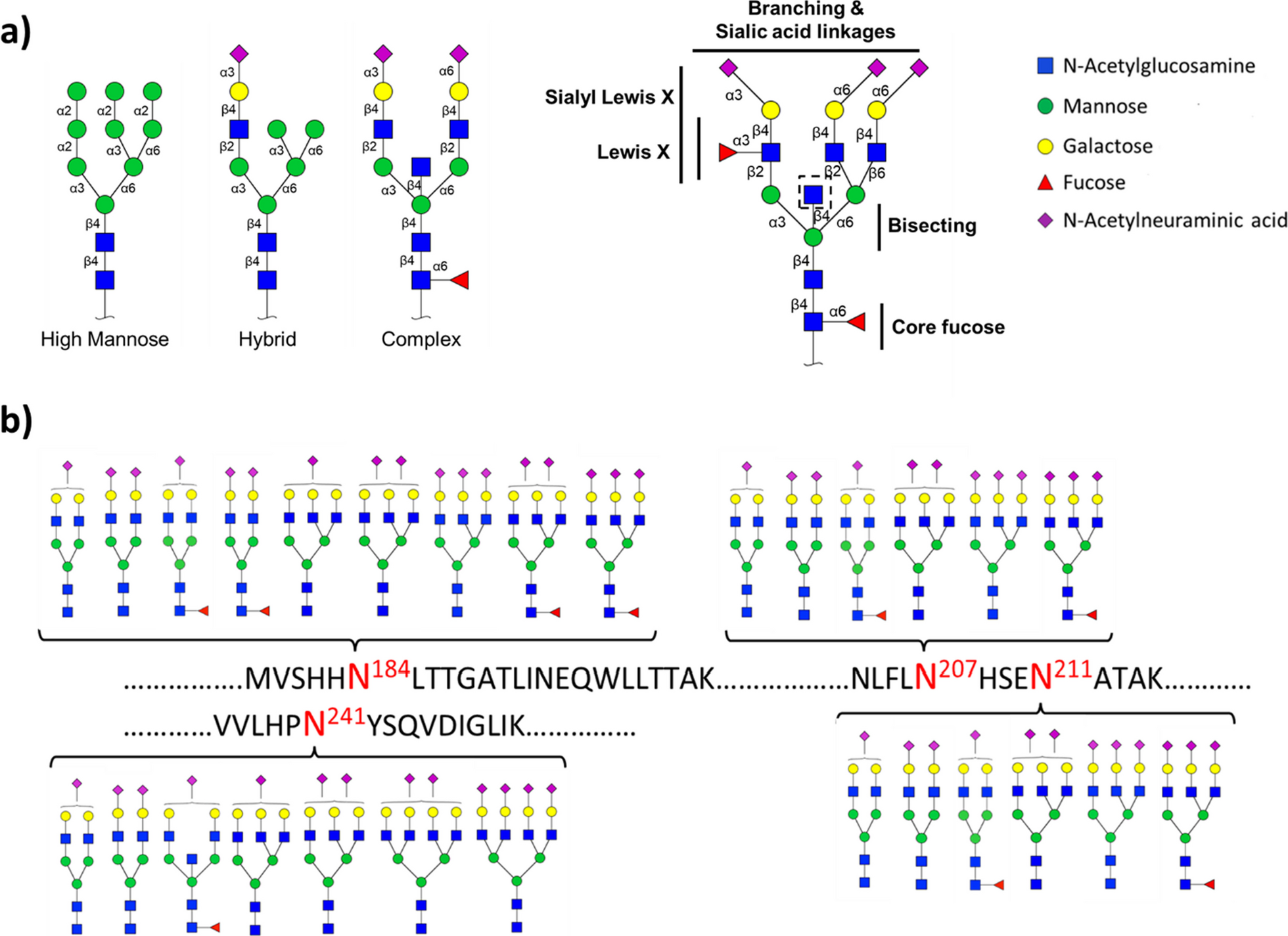


Chemical carcinogen DEN-induced hepatocarcinogenesis goes through premalignant liver cancer progenitor HcPCs to fully established HCC, and we first examined RIG-I expression during hepatocarcinogenesis, including normal hepatocytes, nonaggregated hepatocytes and aggregates containing HcPCs from mice five months post-DEN injection [4, 25], and established HCC cells eight months post-DEN injection. Both protein and mRNA levels of RIG-I were determined to be decreased in HcPCs and established HCC cells, while its levels in normal hepatocytes and nonaggregated hepatocytes were similar (Fig. 1a, b). Thus, RIG-I expression is decreased in the stages from premalignant HcPCs to established HCC during hepatocarcinogenesis, suggesting its potential role in the development of HcPCs.
Fig. 1
IL-6-induced RIG-I decrease in HcPCs promotes hepatocarcinogenesis. a, b RIG-I expression in isolated normal hepatocytes, nonaggregated hepatocytes and HcPCs from male mice five months post-DEN injection, and established HCC cells eight months post-DEN injection was examined by Western blot (a) and qRT-PCR (b, n = 4, one-way ANOVA and Tukey’s multiple comparisons test). c RIG-I expression was examined in male mouse livers in the indicated time periods post continuous intraperitoneal IL-6 injection twice a week. d RIG-I expression was examined in liver tissues of IL-6rahep−/− mice treated as in c. e RIG-I expression was examined in nonaggregate hepatocytes and HcPCs isolated from male IL-6−/− or IL-6rahep−/− mice five months post-DEN injection. f RIG-I expression in liver tissues and isolated hepatocytes from Rig-Ihep−/− mice was confirmed by Western blot. g Representative livers of DEN-induced HCC in male Rig-If/f and Rig-Ihep−/− mice. h Tumor incidence (chi-square test), number and maximum diameter (unpaired t-test) in g were analyzed (n = 12). Data are shown as mean ± s.d. or photographs from one representative of three independent experiments. *P < 0.05, **P < 0.01
The mechanism responsible for the decreased RIG-I in HcPCs was then investigated. As the initiated hepatocytes acquire the ability to autocrine IL-6 thus becoming HcPCs during hepatocarcinogenesis [4], and knockout of IL-6 abolishes inflammation-induced HCC [26], we analyzed whether IL-6 mediated the decrease in hepatic RIG-I. By continuous injection of IL-6 twice a week in vivo, we found that hepatic RIG-I expression was increased in the early phase (within one week) while decreased in the late phase (at two months) (Fig. 1c). In hepatocyte-specific IL-6 receptor knockout IL-6rahep−/− mice, continuous IL-6 injection failed to decrease hepatic RIG-I expression (Fig. 1d). Moreover, in IL-6−/− or IL-6rahep−/− mice, RIG-I expression in isolated HcPCs five months post-DEN injection was not decreased as compared to that in nonaggregated hepatocytes (Fig. 1e). Together, although IL-6 increases hepatic RIG-I expression in the early phase, its expression is decreased in HcPCs, which is mediated by continuous IL-6 stimulation in the late phase.
Decreased hepatic RIG-I expression promotes DEN-induced hepatocarcinogenesis.To analyze the role of RIG-I decrease in hepatocarcinogenesis, we constructed hepatocyte-specific RIG-I knockout Rig-Ihep−/− mouse (Fig. 1f, Additional file 2: Fig. S1a), and found that DEN-induced hepatocarcinogenesis was markedly promoted by hepatic RIG-I deficiency, including increased tumor incidence, number, diameter, and shortened survival (Fig. 1g, h, Additional file 2: Fig. S1b). The DEN plus CCl4-induced hepatocarcinogenesis model was applied, and it was also promoted by hepatic RIG-I deficiency (Additional file 2: Fig. S1c, d). Moreover, to exclude the potential effect of RIG-I as a viral RNA sensor in innate immune cells, we generated Rig-IMac−/− mice and determined that RIG-I in macrophages could not significantly influence DEN-induced hepatocarcinogenesis (Additional file 2: Fig. S1e). Together, hepatic RIG-I deficiency promotes DEN-induced HCC, suggesting that decreased RIG-I expression in HcPCs may participate in hepatocarcinogenesis.
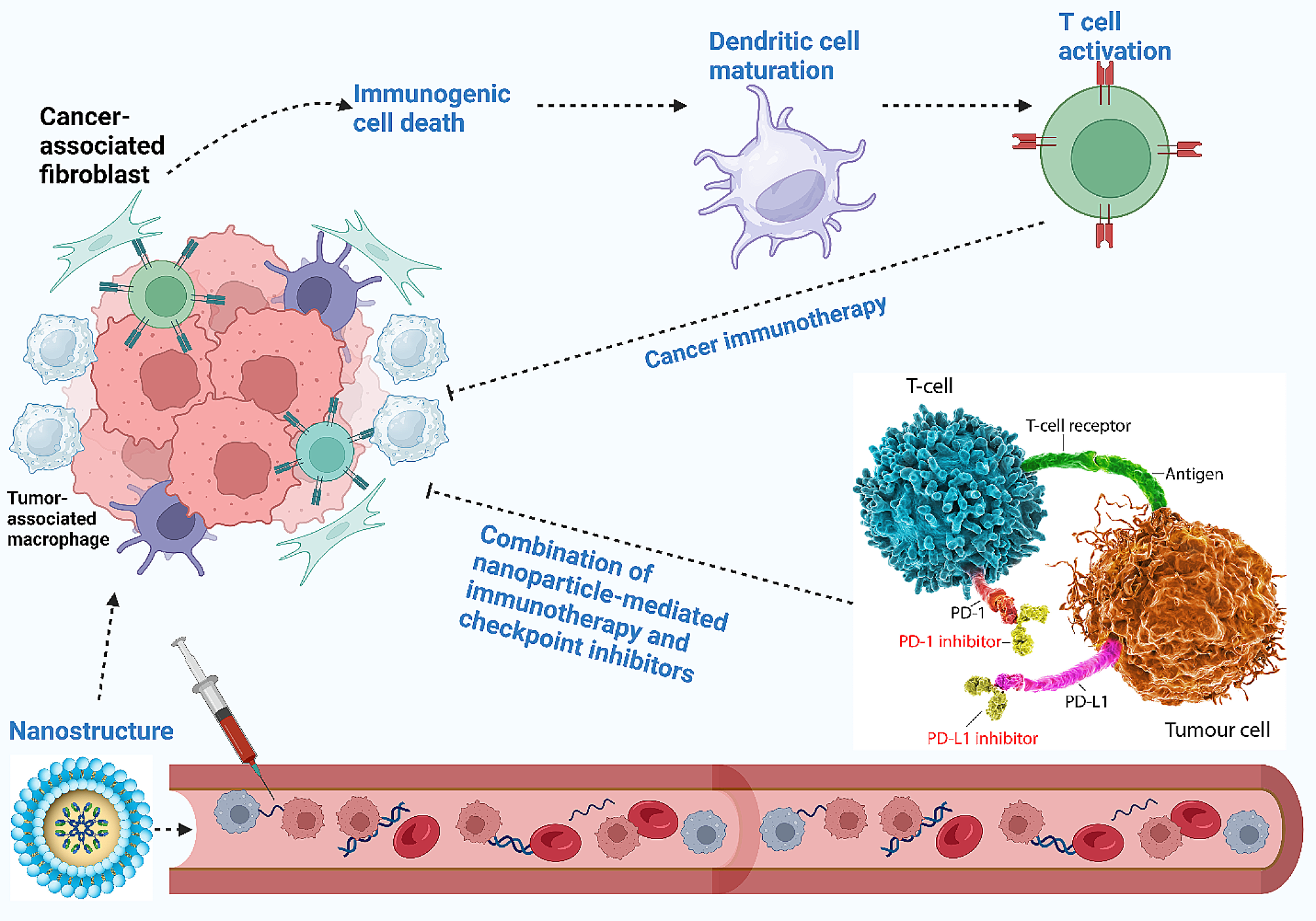
Decreased RIG-I expression in liver cancer progenitor HcPCs promotes their response to IL-6, which viciously drives their progression to established HCCWe next examined the underlying mechanism responsible for Rig-Ihep−/−-promoted hepatocarcinogenesis. The DNA damage, hepatocyte apoptosis, and serum ALT and AST representing liver damage following acute DEN injection were analyzed in Rig-Ihep−/− mice, and DEN-induced liver damage was not influenced by hepatic RIG-I deficiency (Additional file 2: Fig. S2a–e). The DEN-induced hepatic production of proinflammatory cytokines including IL-6 and TNF-α, infiltration of leukocytes, and compensatory hepatocyte proliferation were also not significantly influenced by Rig-Ihep−/− (Additional file 2: Fig. S2f–h). We then performed protein phosphorylation microarray to elucidate the altered intracellular signaling in Rig-Ihep−/− liver following acute DEN injection, and found that STAT3 phosphorylation was the most increased by hepatic RIG-I deficiency (Additional file 2: Fig. S2i). The increased DEN-induced STAT3 phosphorylation was then confirmed in Rig-Ihep−/− liver (Fig. 2a), suggesting that RIG-I deficiency may enhance STAT3 activation to promote hepatocarcinogenesis.
Fig. 2
Decreased RIG-I in liver cancer progenitor HcPCs promotes their response to IL-6, which viciously drives their progression to established HCC. a STAT3 phosphorylation and activation were examined in liver tissues from male Rig-If/f and Rig-Ihep−/− mice 48 h post-DEN administration. b Tumor incidence (chi-square test), number and maximum diameter (unpaired t-test) of DEN-induced HCC in male Rig-If/fIL-6−/− and Rig-Ihep−/−IL-6−/− mice were analyzed (n = 12). c Tumor incidence (chi-square test), number and maximum diameter (unpaired t-test) of DEN-induced HCC in male IL-6rahep−/− and Rig-Ihep−/−IL-6rahep−/− mice were analyzed (n = 12). d IL-6-induced STAT3 phosphorylation in the indicated time periods was examined in liver tissues and isolated hepatocytes from male Rig-If/f and Rig-Ihep−/− mice. e IL-6-induced Saa1 mRNA expression in the indicated time periods was examined in liver tissues and isolated hepatocytes from male Rig-If/f and Rig-Ihep−/− mice (n = 4, unpaired t-test). f Isolated nonaggregated hepatocytes and HcPCs were stimulated with IL-6 for the indicated time periods, and STAT3 phosphorylation was examined. g Isolated nonaggregated hepatocytes and HcPCs from control AAV8 or AAV8-RIG-I-treated male mice were stimulated as in f, and STAT3 phosphorylation was examined. h Isolated HcPCs from male Rig-If/f and Rig-Ihep−/− mice were stimulated with IL-6 for 30 min, and STAT3 phosphorylation was examined. i Tumor number and maximum diameter were analyzed in male mice transplanted by intrasplenic injection with equal amounts of isolated HcPCs from Rig-If/f and Rig-Ihep−/− mice (n = 6, unpaired t-test). Data are shown as mean ± s.d. or photographs from one representative of three independent experiments. *P < 0.05, **P < 0.01, ▲P > 0.05
As IL-6 effects mainly through Janus kinase 2 (JAK2)-STAT3 signaling and is the key proinflammatory cytokine driving hepatocarcinogenesis [26, 27], we examined whether hepatic RIG-I deficiency-promoted hepatocarcinogenesis was dependent on IL-6-STAT3 signaling. Rig-Ihep−/−IL-6−/− double knockout (DKO) mice and Rig-Ihep−/−IL-6rahep−/− DKO mice were generated, respectively. Loss of IL-6 diminished DEN-induced HCC, and importantly, Rig-Ihep−/−IL-6−/− mice and IL-6−/− mice displayed a similar reduced induction of HCC by DEN (Fig. 2b), suggesting that hepatic RIG-I deficiency failed to promote hepatocarcinogenesis under IL-6 deficiency. Similar results were obtained in Rig-Ihep−/−IL-6rahep−/− DKO mice (Fig. 2c). Therefore, hepatocyte-specific RIG-I deficiency-promoted hepatocarcinogenesis is dependent on the proinflammatory cytokine IL-6.
DEN-induced production of hepatic IL-6 was not influenced by RIG-I deficiency (Additional file 2: Fig. S2f), whether hepatic RIG-I deficiency promotes hepatocarcinogenesis by enhancing IL-6-STAT3 effector signaling was then examined. We found that IL-6-induced STAT3 phosphorylation was significantly promoted by hepatic RIG-I deficiency, both upon IL-6 injection in vivo and IL-6 stimulation in primary hepatocytes in vitro (Fig. 2d). The expression of IL-6-induced downstream gene Saa1 was also enhanced by hepatic RIG-I deficiency (Fig. 2e). Thus, hepatic RIG-I deficiency-promoted hepatocarcinogenesis is mediated by the enhanced oncogenic IL-6-STAT3 effector signaling.
Since RIG-I expression is decreased in liver cancer progenitor HcPCs and RIG-I deficiency promotes IL-6-driven hepatocarcinogenesis, we presumed that not only IL-6 was autocrined by HcPCs, but also their response to IL-6 was enhanced. IL-6-induced STAT3 activation was significantly enhanced in HcPCs as compared to that in nonaggregated hepatocytes (Fig. 2f). We also rescued RIG-I expression in HcPCs using AAV8-mediated gene delivery, and confirmed that IL-6-STAT3 signaling was compromised, to the level similar with that in nonaggregated hepatocytes of control mice (Fig. 2g). Hence, the response to IL-6 is enhanced in HcPCs, which is mediated by the decreased RIG-I expression.
To confirm that decreased RIG-I in HcPCs promotes hepatocarcinogenesis, especially in the stages from HcPCs to established HCC, we isolated CD44+ HcPCs from the livers of Rig-If/f and Rig-Ihep−/− mice five months post-DEN injection, and determined that IL-6-induced STAT3 activation was enhanced by RIG-I deficiency in HcPCs (Fig. 2h). Equal amounts of HcPCs from Rig-If/f or Rig-Ihep−/− were transplanted into wildtype mice, which were then injected weekly with CCl4 to induce liver inflammation for five months to drive the transplanted HcPCs to established HCC [4]. RIG-I deficient HcPCs generated more and larger HCC nodules as compared to those of HcPCs from RIG-If/f mice (Fig. 2i), suggesting that decreased RIG-I in HcPCs promotes their progression to HCC. Altogether, we conclude that IL-6-induced RIG-I decrease and decreased RIG-I-enhanced IL-6 response in HcPCs may cause vicious feedforward progression from premalignant HcPCs to fully established HCC.
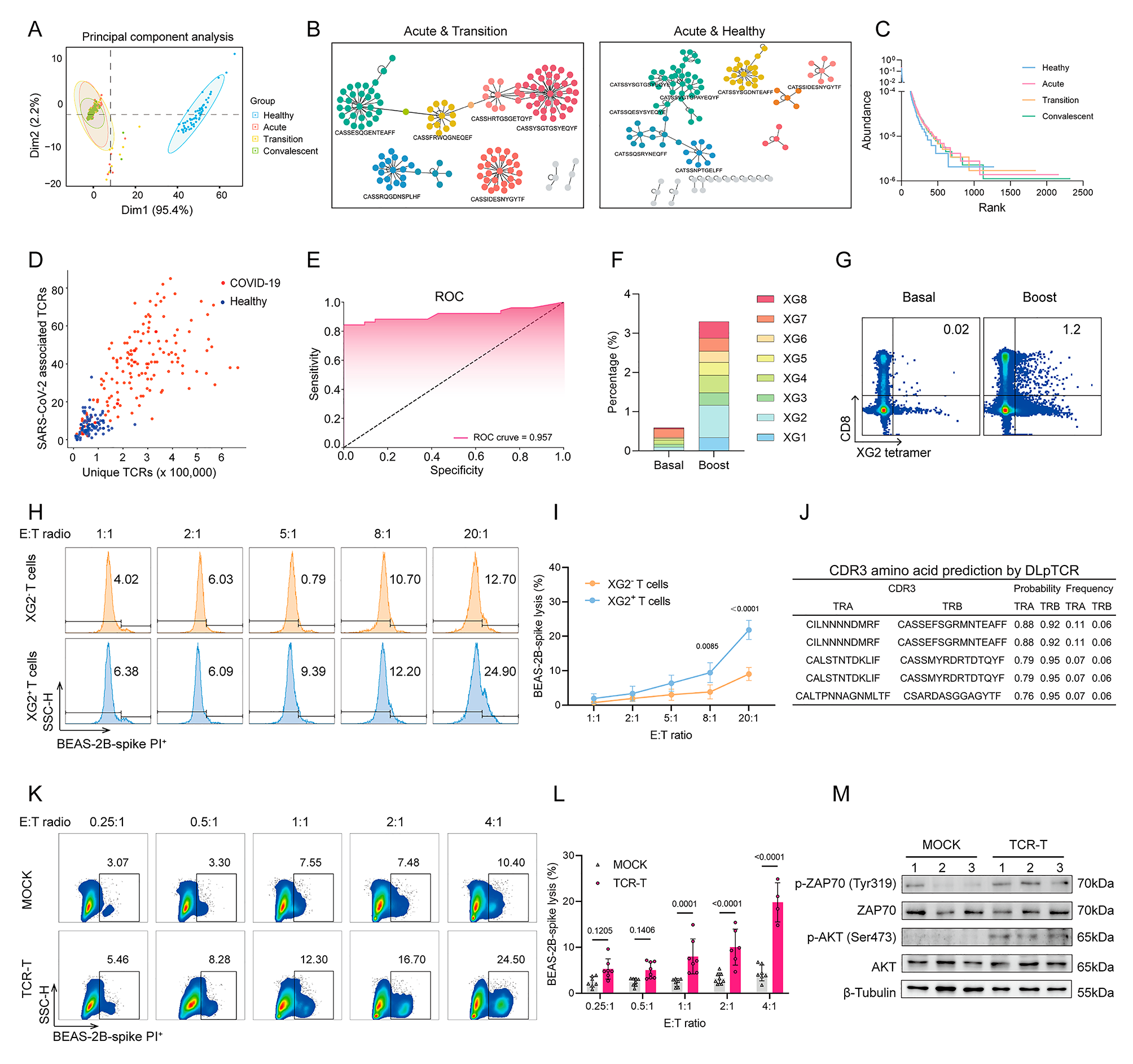
RIG-I associates with STAT3 to impede JAK2-STAT3 interaction and inhibit IL-6 effector signaling.The mechanism responsible for RIG-I-mediated inhibition of IL-6-STAT3 signaling was then examined. We found that IL-6 could induce the association between RIG-I and STAT3, both upon IL-6 injection in vivo and IL-6 stimulation in primary hepatocytes in vitro (Fig. 3a, Additional file 2: Fig. S3a). To determine how RIG-I binding to STAT3 suppresses STAT3 activation, we screened the kinases and regulators involved in IL-6-STAT3 signaling, and found that STAT3 could bind to JAK1, JAK2, SHP1, SHP2, and SOCS3, but not SOCS1, PIAS1, or PIAS3, upon IL-6 stimulation in the liver (Additional file 2: Fig. S3b). Among them, IL-6-induced JAK2-STAT3 interaction was significantly enhanced by hepatic RIG-I deficiency, both by IL-6 injection in vivo and IL-6 stimulation in primary hepatocytes in vitro (Fig. 3b, Additional file 2: Fig. S3c), while IL-6-induced JAK2 phosphorylation was not influenced (Additional file 2: Fig. S3d). As JAK2-phosphorylated STAT3 is the key pathway in IL-6 effects, and RIG-I deficiency failed to promote IL-6-STAT3 activation under inhibition of JAK2 (Additional file 2: Fig. S3e), hepatic RIG-I deficiency-promoted STAT3 activation is mediated by the enhanced JAK2-STAT3 interaction. Moreover, the tagged truncates of RIG-I, JAK2, and STAT3 were constructed, and both CARD domain of RIG-I and JH2 domain of JAK2 could bind the SH2-TA domain of STAT3, which contains the phosphorylation site tyrosine 705 (Additional file 2: Fig. S3f). Thus, RIG-I associates with STAT3 to impede JAK2-STAT3 interaction and inhibit IL-6 effector signaling. Together with the IL-6-induced hepatic RIG-I expression in the early phase (Fig. 1c), all these data determine that IL-6 induces RIG-I-STAT3 association and RIG-I expression to feedback inhibit JAK2-STAT3 interaction and IL-6 effector signaling in the early phase.
Fig. 3
IL-6 induces RIG-I demethylation to enhance RIG-I-STAT3 association and feedback impede JAK2-STAT3 interaction. a RIG-I-STAT3 association induced by IL-6 was examined by immunoprecipitation in male mouse liver tissues. b IL-6-induced JAK2-STAT3 interaction was evaluated by immunoprecipitation in liver tissues from male Rig-If/f and Rig-Ihep−/− mice. c, d V5-tagged STAT3 and Flag-tagged RIG-I mutants as indicated were transfected into HHL5 hepatocyte cell line, and their association was tested by immunoprecipitation. e Methylated RIG-I at K18 and K146 were examined by the specific antibodies in the precipitates by total RIG-I antibody from the liver tissues upon IL-6 stimulation. f IL-6-induced STAT3 phosphorylation was evaluated in liver tissues from wildtype, RIG-I K18M+K146M or K18A+K146A mutant male mice upon IL-6 stimulation. g IL-6-induced RIG-I-STAT3 association and JAK2-STAT3 interaction were examined using immunoprecipitation in the liver tissues from male wildtype, RIG-I K18M+K146M or K18A+K146A mutant mice upon IL-6 stimulation. h Tumor incidence (chi-square test), number and maximum diameter (unpaired t-test) of DEN-induced HCC in male wildtype, RIG-I K18M+K146M or K18A+K146A mutant mice were analyzed (n = 12). Data are shown as mean ± s.d. or photographs from one representative of three independent experiments. *P < 0.05, **P < 0.01
IL-6 induces the demethylation of RIG-I to enhance RIG-I-STAT3 association.We next examined the mechanism responsible for IL-6-induced RIG-I-STAT3 association. As protein PTM plays critical roles in protein–protein interaction, the PTM of RIG-I following IL-6 stimulation was analyzed by mass spectrometry, especially in the CARD domain which is responsible for RIG-I-STAT3 association. Methylation, acetylation, and phosphorylation of RIG-I were screened. Mono-methylation at K18, K146, and acetylation at K95, K172 were identified in untreated, while acetylation at K59, K154 were identified in IL-6-stimulated hepatocytes, and mono-methylation at K48 and phosphorylation at S8, T55 were identified in both. The mutants K48A, K59A, K95A, K154A, K172A, S8A, and T55A had no effect on the RIG-I-STAT3 association (Additional file 2: Fig. S3g). However, the mutants K18M and K146M mimicking mono-methylation of evolutionarily conserved K18 and K146 both significantly decreased RIG-I-STAT3 association, and K18M + K146M mutant abolished the association (Fig. 3c, Additional file 2: Fig. S3h, i). The K18A, K146A, K18R, K146R mutants of RIG-I mimicking demethylation were also constructed, and their association with STAT3 were significantly enhanced (Fig. 3d), suggesting that demethylation of RIG-I at both K18 and K146 may be important for RIG-I-STAT3 association. Furthermore, rabbit polyclonal antibodies specifically recognizing the K18 or K146 mono-methylated RIG-I were generated, respectively (Additional file 2: Fig. S3j, k), and IL-6-induced RIG-I demethylation at these sites was confirmed both in liver tissues in vivo and in primary hepatocytes in vitro (Fig. 3e, Additional file 2: Fig. S3l). Thus, IL-6-induced RIG-I-STAT3 association was dependent on the induced demethylation at K18 and K146 of hepatic RIG-I.
To confirm the role of demethylated RIG-I in associating and suppressing IL-6-induced STAT3 activation, we constructed the K18M+K146M mutant mouse mimicking mono-methylated RIG-I, and K18A+K146A mutant mouse mimicking demethylated RIG-I (Additional file 2: Fig. S3m). IL-6-induced hepatic STAT3 phosphorylation was significantly increased in K18M+K146M mice, while decreased in K18A+K146A mice, both by IL-6 injection in vivo and IL-6 stimulation in primary hepatocytes in vitro (Fig. 3f and Additional file 2: Fig. S3n). Similarly, hepatic RIG-I-STAT3 association was markedly decreased in K18M+K146M mice, while increased in K18A+K146A mice; IL-6-induced JAK2-STAT3 association was enhanced in K18M+K146M mice, while decreased in K18A+K146A mice (Fig. 3g). Furthermore, DEN-induced hepatocarcinogenesis was increased in K18M+K146M mice, while decreased in K18A+K146A mice (Fig. 3h). Together, we conclude that IL-6-induced RIG-I demethylation at K18 and K146 feedback associates with STAT3 and inhibits IL-6-STAT3 effector signaling.
Demethylase JMJD4 is responsible for IL-6-induced demethylation of RIG-I.The mechanism responsible for IL-6-induced RIG-I demethylation was then investigated. We immunoprecipitated Flag-tagged RIG-I from lysates of IL-6-stimulated HHL5 hepatocyte cell line, then used mass spectrometry to identify RIG-I-associated proteins, and selected JMJD4 as the candidate because of its demethylase activity (Additional file 2: Fig. S4a). The IL-6-induced JMJD4-RIG-I association was confirmed by immunoprecipitation (Fig. 4a). The tagged truncates were constructed, and JMJC demethylase catalytic domain of JMJD4 also associated with the CARD domain of RIG-I, where K18 and K146 locate (Additional file 2: Fig. S4b). Thus, demethylase JMJD4 associates with RIG-I following IL-6 stimulation, which may participate in the IL-6-induced RIG-I demethylation.
Fig. 4
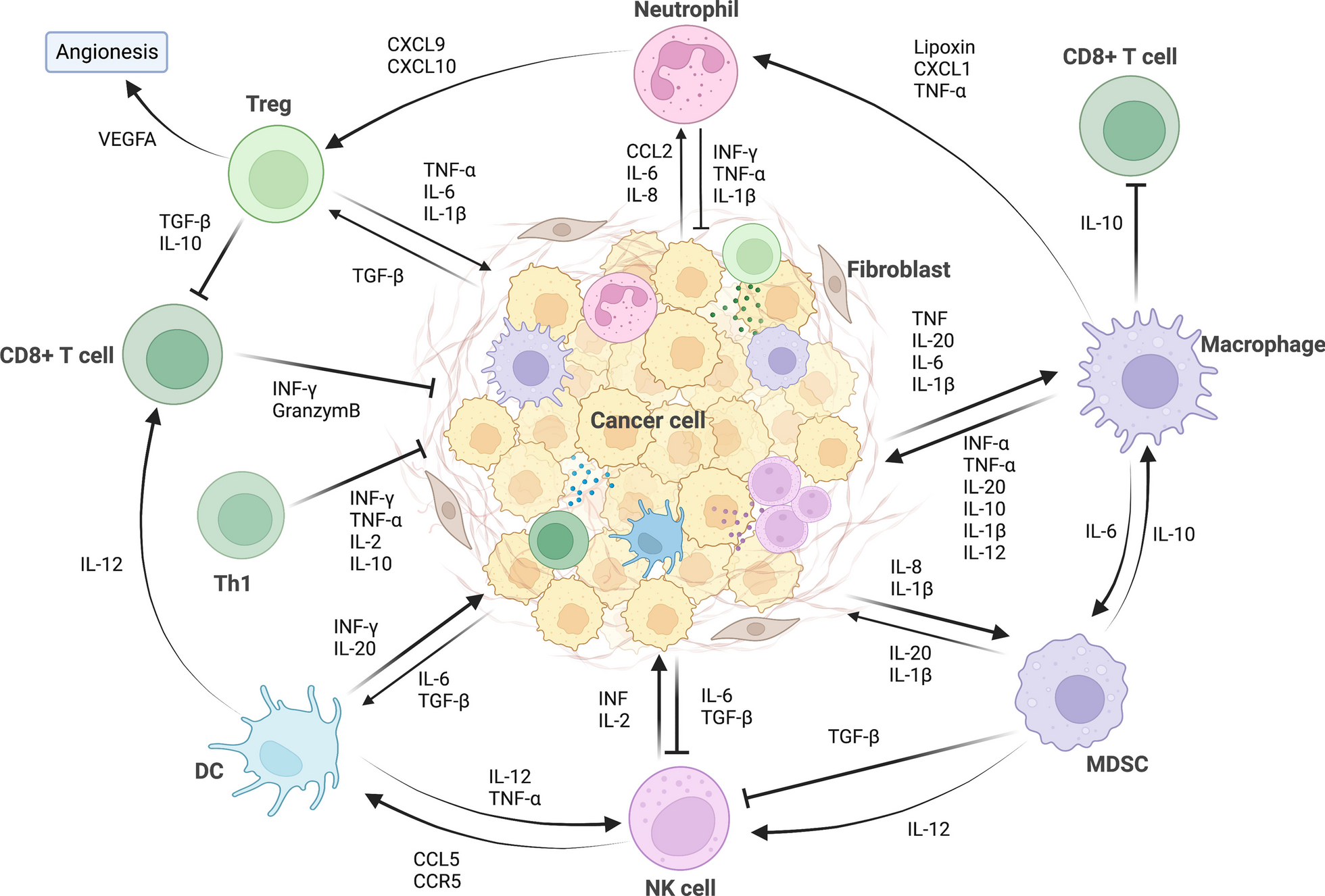
JMJD4-demethylated RIG-I inhibits DEN-induced hepatocarcinogenesis. a JMJD4-RIG-I association induced by IL-6 was examined by immunoprecipitation in male mouse liver tissues. b Methylated RIG-I at K18 and K146 upon IL-6 stimulation was examined in male Jmjd4f/f and Jmjd4hep−/− liver tissues and isolated primary hepatocytes as indicated. c IL-6-induced RIG-I-STAT3 association was evaluated by immunoprecipitation in liver tissues from male Jmjd4f/f and Jmjd4hep−/− mice. d IL-6-induced STAT3 phosphorylation was evaluated in liver tissues from male Jmjd4f/f and Jmjd4hep−/− mice upon IL-6 stimulation. e Tumor incidence (chi-square test), number and maximum diameter (unpaired t-test) of DEN-induced HCC in Jmjd4f/f and Jmjd4hep−/− mice were analyzed (n = 12). f JMJD4 expression in isolated normal hepatocytes, nonaggregated hepatocytes and HcPCs from male mice five months post-DEN injection, and established HCC cells eight months post-DEN injection was examined by Western blot. g Tumor incidence (chi-square test), number and maximum diameter (unpaired t-test) of DEN-induced HCC in male Jmjd4f/fRig-If/f, Rig-Ihep−/− and Jmjd4hep−/−Rig-Ihep−/− mice, or in male Jmjd4f/f, Jmjd4f/f RIG-I K18A+K146A and Jmjd4hep−/− RIG-I K18A+K146A mice as indicated were analyzed (n = 12). h IL-6-induced STAT3 phosphorylation was evaluated in liver tissues from male Jmjd4f/fRig-If/f, Rig-Ihep−/− and Jmjd4hep−/−Rig-Ihep−/− mice upon IL-6 stimulation. Data are shown as mean ± s.d. or photographs from one representative of three independent experiments. *P < 0.05, **P < 0.01, ▲P > 0.05
We next constructed hepatocyte-specific JMJD4 knockout mouse (Additional file 2: Fig. S4c, d), and determined that IL-6-induced hepatic RIG-I demethylation at K18 and K146 was abolished by JMJD4 deficiency, both by IL-6 injection in vivo and IL-6 stimulation in primary hepatocytes in vitro (Fig. 4b). IL-6-induced RIG-I-STAT3 association was markedly decreased while STAT3 phosphorylation was enhanced by hepatic JMJD4 deficiency (Fig. 4c, d). Together, these data determine that IL-6 induces JMJD4-erased methylation of RIG-I to enhance RIG-I-STAT3 association and feedback inhibit IL-6-STAT3 signaling.
JMJD4 and RIG-I co-operatively suppress DEN-induced hepatocarcinogenesis.As JMJD4-erased methylation of RIG-I feedback suppresses IL-6-STAT3 effector signaling, we further examined the role of JMJD4 in DEN-induced hepatocarcinogenesis. Hepatocyte-specific JMJD4 knockout significantly promoted both DEN and DEN plus CCl4-induced hepatocarcinogenesis (Fig. 4e, Additional file 2: Fig. S4e), which is similar to those in Rig-Ihep−/− mice. JMJD4 expression was also analyzed in mouse HcPCs and HCC cells, and its expression moderately decreased as compared to that in normal hepatocytes and nonaggregates (Fig. 4f, Additional file 2: Fig. S4f). Together with the data that the markedly decreased RIG-I in HcPCs promotes hepatocarcinogenesis, this potentially decreased JMJD4 may further enhance IL-6 response and the corresponding hepatocarcinogenesis.
The hepatocyte-specific JMJD4 and RIG-I DKO mouse and Jmjd4hep−/− plus RIG-I K18A+K146A mouse were then generated, respectively, and we found that the DEN-induced hepatocarcinogenesis in Jmjd4hep−/−Rig-Ihep−/− mice was similar to that in Rig-Ihep−/− mice, and hepatocarcinogenesis in Jmjd4hep−/− plus RIG-I K18A+K146A mice was similar to that in Jmjd4f/f plus RIG-I K18A+K146A mouse (Fig. 4g), suggesting that JMJD4 deficiency-promoted hepatocarcinogenesis is dependent on RIG-I and its demethylation. Moreover, IL-6-induced hepatic STAT3 phosphorylation was enhanced in Jmjd4hep−/−Rig-Ihep−/− mice, which was similar to that in Rig-Ihep−/− mice (Fig. 4h). Thus, IL-6-induced hepatic JMJD4-RIG-I association co-operatively feedback suppresses IL-6-STAT3 effector signaling and DEN-induced hepatocarcinogenesis. Altogether, we conclude that decreased RIG-I in liver cancer progenitor HcPCs promotes their response to IL-6, which drives the progression from HcPCs to fully established HCC in the DEN model mimicking necroinflammation-induced hepatocarcinogenesis.
Opposite to DEN model, Rig-I hep−/− suppressed NASH-induced HCCAlthough the DEN-induced hepatocarcinogenesis model has been widely used for its ease and consistency in generating HCC as well as HcPCs, a recent study determined that tumors of Stelic Animal Model (STAM), by using streptozotocin (STZ) and high-fat diet (HFD) to mimic NASH-induced HCC, were the most molecularly similar to human HCC among the available hepatocarcinogenesis mouse models [23]. We thus generated STAM tumors in Rig-If/f and Rig-Ihep−/− mice. Interestingly, hepatocyte-specific RIG-I deficiency nearly abolished NASH-induced hepatocarcinogenesis, suggesting the tumor-promotive role of RIG-I in NASH-induced HCC, which is opposite to the tumor-suppressive role of RIG-I in the DEN model (Fig. 5a, b, Additional file 2: Fig. S5a, b). In another NASH-induced HCC mouse model that western diet (WD) plus CCl4 injection, hepatic RIG-I deficiency also markedly inhibited hepatocarcinogenesis (Additional file 2: Fig. S5c). The generated STAM tumors of Rig-If/f and Rig-Ihep−/− mice were then analyzed. Although the pathological features of tumors were similar, we interestingly found that the NASH features in nontumor liver tissues, including ballooning and inflammation, were induced in Rig-If/f mice, while abolished by hepatic RIG-I deficiency (Fig. 5c). Moreover, hepatic oil red O staining also determined that lipid accumulation was abolished by RIG-I deficiency in STAM model (Fig. 5d). Thus, these data indicate that hepatic RIG-I deficiency may suppress NASH progression, and then NASH-induced hepatocarcinogenesis.
Fig. 5
Hepatocyte-specific RIG-I deficiency abolishes steatosis, and the following NASH, and NASH-induced hepatocarcinogenesis. a Tumor incidence (chi-square test), number and maximum diameter (unpaired t-test) of STAM HCC in male Rig-If/f and Rig-Ihep−/− mice were analyzed (n = 12). b Representative livers of STAM HCC in male Rig-If/f and Rig-Ihep−/− mice. c, d HE (c) and oil red O (d) staining were analyzed in nontumor liver tissues of STAM model in male Rig-If/f and Rig-Ihep−/− mice. Scale bars: 20 μm. e, f HE (e) and oil red O (f) staining were analyzed in liver tissues of MCD model in male Rig-If/f and Rig-Ihep−/− mice. Scale bars: 20 μm. g, h HE (g) and oil red O (h) staining were analyzed in liver tissues of HFD model in male Rig-If/f and Rig-Ihep−/− mice. Scale bars: 20 μm. i, j Hepatic TG and TC (i), and serum TG and TC (j) were examined in HFD-treated male Rig-If/f and Rig-Ihep−/− mice (n = 4, unpaired t-test). Data are shown as mean ± s.d. or photographs from one representative of three independent experiments. *P < 0.05, **P < 0.01, ▲P > 0.05
The methionine and choline deficient diet (MCD) and choline deficient-high-fat diet (CD-HFD) were then used to induce NASH in Rig-If/f and Rig-Ihep−/− mice to confirm the inhibited NASH progression by hepatic RIG-I deficiency. We found that NASH progression, the induced hepatic proinflammatory IL-6, and the fibrosis feature were all markedly suppressed in Rig-Ihep−/− livers of MCD model (Fig. 5e, Additional file 2: Fig. S5d, e, f), and NASH features were also abolished in Rig-Ihep−/− mice of CD-HFD model (Additional file 2: Fig. S5g). Furthermore, hepatic oil red O staining confirmed that lipid accumulation was inhibited by RIG-I deficiency in these NASH models (Fig. 5f, Additional file 2: Fig. S5h). Hence, we conclude that hepatic RIG-I deficiency suppresses NASH and NASH-induced HCC, and the tumor-promotive role of RIG-I in NASH-induced HCC is opposite to the tumor-suppressive role of RIG-I in DEN-induced hepatocarcinogenesis.
Hepatic RIG-I deficiency suppressed the occurrence of steatosis, especially by the diminished hepatic cholesterol accumulationAs NASH-induced HCC goes through steatosis, NASH, fibrosis, cirrhosis, and HCC, we next examined whether hepatic RIG-I deficiency could inhibit the development of the first step steatosis. Using HFD to induce simple steatosis but minor NASH, we found that hepatic RIG-I deficiency suppressed HFD-induced steatosis as compared to that in Rig-If/f mice (Fig. 5g, Additional file 2: Fig. S5i). Hepatic oil red O staining also determined that lipid accumulation was abolished by RIG-I deficiency in the HFD model (Fig. 5h). The increase in body and liver weight induced by HFD was also suppressed in Rig-Ihep−/− mice (Additional file 2: Fig. S5j). Thus, hepatic RIG-I deficiency inhibits the occurrence of the first step steatosis, which then abolishes the following NASH and NASH-induced HCC. Moreover, we examined RIG-I expression in the liver of HFD-induced steatosis, and found the increased RIG-I in the steatosis liver (Additional file 2: Fig. S5k). Together, RIG-I expression is increased by hepatic steatosis, and RIG-I deficiency in hepatocytes abolishes steatosis development.
The accumulation of triglyceride (TG) and total cholesterol (TC) in hepatocytes is the main feature of hepatic steatosis, and they were then examined. In HFD-treated mice, both hepatic TG and TC were markedly suppressed in Rig-Ihep−/− mice (Fig. 5i). Remarkably, hepatic TC was equivalent between ND and HFD groups of Rig-Ihep−/− mice, suggesting the most markedly suppressed hepatic TC by RIG-I deficiency, and HFD could not increase hepatic cholesterol in Rig-Ihep−/− mice (Fig. 5i). Furthermore, the increased serum cholesterol in HFD-treated mice was abolished by hepatic RIG-I deficiency, while serum TG was not significantly influenced (Fig. 5j). Hence, RIG-I deficiency in hepatocytes abolished the HFD-induced increase in both hepatic and serum cholesterol, which may be responsible for the abolished steatosis in Rig-Ihep−/− mice. As atherosclerosis (AS) is the most threatening extrahepatic disease mediated by the increased serum cholesterol during hepatic steatosis, we also examined whether RIG-I deficiency could inhibit AS progression. Using the AS model of HFD-treated ApoE knockout mouse, we found that RIG-I deficiency markedly suppressed AS progression (Additional file 2: Fig. S5l, m). Thus, we conclude that RIG-I deficiency abolishes hepatic cholesterol accumulation to suppress both hepatic steatosis and high serum cholesterol-mediated extrahepatic disease.
As RIG-I expression is increased in the steatosis liver, we then examined its expression in NASH livers, and found that RIG-I expression was decreased in both NASH livers and NASH-induced HCC tissues (Additional file 2: Fig. S5n). Considering liver injury and inflammation are the typical features of NASH as compared to hepatic simple steatosis, and IL-6 is determined to decrease hepatic RIG-I expression (Fig. 1c, d), we also found that RIG-I decrease in NASH was suppressed by IL-6 knockout or hepatic IL-6 receptor knockout, thus suggesting that proinflammatory cytokine IL-6 mediates RIG-I decrease in NASH (Additional file 2: Fig. S5o). Together, we conclude that although hepatic RIG-I expression is increased in the first step steatosis stage to promote lipid accumulation and steatosis development, its expression is decreased in the following NASH and NASH-induced HCC, which is mediated by the proinflammatory cytokine IL-6.
Methylated RIG-I associates AMPKα to inhibit HMGCR phosphorylation, thus increasing HMGCR enzymatic activity and enhancing cholesterol synthesisTo elucidate the mechanism responsible for RIG-I deficiency-mediated inhibition of cholesterol accumulation and steatosis, we performed the transcriptome analysis between the livers of Rig-If/f and Rig-Ihep−/− mice. As the different hepatic lipid accumulation following HFD treatment in Rig-If/f and Rig-Ihep−/− mice may influence the expression of the metabolic enzymes, we chose the livers under normal diet for analysis. The mRNAs of genes involved in lipid uptake, synthesis, transport, and excretion were not significantly influenced by hepatic RIG-I deficiency (Additional file 2: Fig. S6a). We then presumed that the protein levels of the cholesterol metabolic genes and/or their phosphorylation might be modulated by hepatic RIG-I deficiency, and screened them using Western blot. Among them, the phosphorylation of HMGCR, the rate-limiting enzyme for cholesterol synthesis and its phosphorylation leads to inactivation [28], was the most markedly increased in Rig-Ihep−/− livers (Fig. 6a). Thus, the phosphorylation of HMGCR, leading to the inactivation of cholesterol synthesis, is increased by hepatic RIG-I deficiency, which may be responsible for the most significantly suppressed cholesterol accumulation and hepatic steatosis in Rig-Ihep−/− mice.
Fig. 6
Methylated RIG-I associates AMPKα to inhibit HMGCR phosphorylation and enhance cholesterol synthesis. a HMGCR, ACC, AMPKα, and their phosphorylation were examined in liver tissues of male Rig-If/f and Rig-Ihep−/− mice. b HMGCR, AMPKα, and their phosphorylation were examined in liver tissues of male Rig-If/f and Rig-Ihep−/− mice fasting for 16 h. c The AMPKα-HMGCR interaction and AMPKα-RIG-I interaction upon fasting for 16 h were evaluated using immunoprecipitation in liver tissues from male Rig-If/f and Rig-Ihep−/− mice. d V5-tagged AMPKα and Flag-tagged RIG-I mutants as indicated were transfected into HHL5 hepatocyte cell line, and their association was tested by immunoprecipitation. e, f HE (e) and oil red O (f) staining were analyzed in the liver tissues of HFD model in male wildtype, RIG-I K18M+K146M or K18A+K146A mutant mice. Scale bars: 20 μm. g, h HE (g) and oil red O (h) staining were analyzed in the liver tissues of HFD model in male Jmjd4f/f and Jmjd4hep−/− mice. Scale bars: 20 μm. Data are shown as photographs from one representative of three independent experiments
As HMGCR phosphorylation and inactivation are mediated by upstream activated kinase AMPKα [28], we next examined the association between AMPKα and HMGCR to analyze whether their association was modulated by RIG-I. In RIG-Ihep−/− livers, both the basal level of HMGCR phosphorylation and its induction by fasting or metformin treatment were significantly enhanced, while the phosphorylation of upstream AMPKα was not influenced (Fig. 6b, Additional file 2: Fig. S6b), suggesting that AMPKα activation is not influenced by RIG-I deficiency. RIG-I was found to constitutively associate with AMPKα, and both the basal level and induced association between AMPKα and downstream HMGCR were significantly enhanced by hepatic RIG-I deficiency (Fig. 6c, Additional file 2: Fig. S6c), determining that AMPKα-HMGCR interaction is inhibited by RIG-I-AMPKα association. Moreover, the N-terminal CARD domain of RIG-I associated with the kinase domain of AMPKα, which was also responsible for the association between AMPKα and C-terminal catalytic domain of HMGCR (Additional file 2: Fig. S6d, e). Together, we conclude that hepatic RIG-I constitutively associates with AMPKα to impede HMGCR, thus inhibiting HMGCR phosphorylation and enhancing its enzymatic activity to synthetize cholesterol.
Since PTM of proteins is important for protein–protein interaction, and methylation at K18, K146, and acetylation at K48, K95, K172 were found to be constitutively existed in RIG-I, we further examined whether these PTMs could influence RIG-I-AMPKα association. The respective mutants at these sites were used, and interestingly, only K18A and K146A mutants mimicking demethylation markedly inhibited RIG-I-AMPKα association, while K18M and K146M mutants mimicking mono-methylation enhanced their association, suggesting that the constitutive K18 and K146 methylation of RIG-I is responsible for its interaction with AMPKα (Fig. 6d). Together with the result that IL-6 induced the demethylation of RIG-I at these two sites, we confirmed that the constitutive RIG-I-AMPKα association was inhibited by IL-6 administration (Additional file
留言 (0)