
記住我
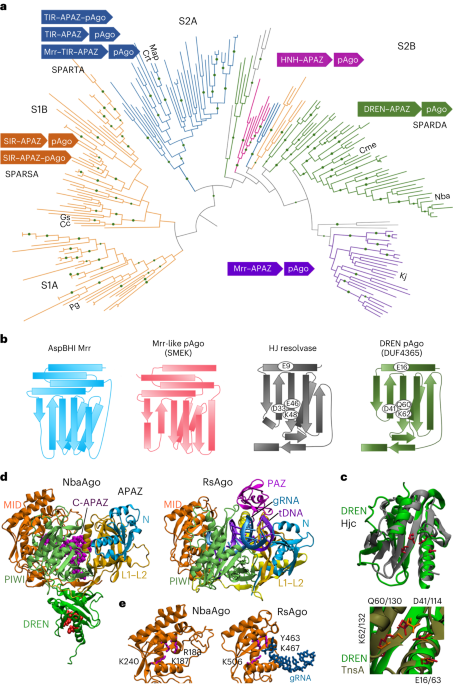
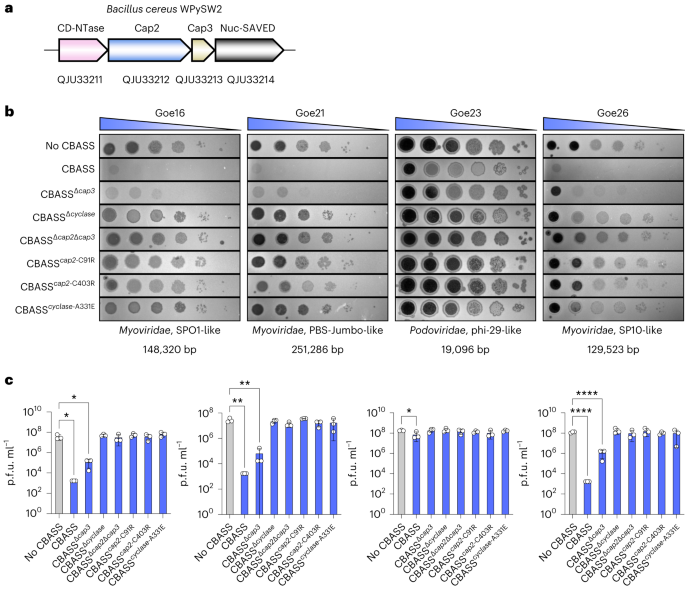
Phages encode diverse anti-defence strategies against the bacterial defence systems they are likely to encounter13,33,39,40, which in turn can render these systems ineffective for either phage immunity or phage engineering. To determine whether Cas13 might be useful as both a broad-spectrum phage defence and a phage genome editing tool, we began by investigating the distribution of Cas13 effectors across bacterial phyla. We performed a bioinformatic search for Cas13 proteins across NCBI and Genome Taxonomy Database (GTDB) genomes, culminating in a non-redundant set of 224 Cas13 protein sequences (Fig. 1 and Supplementary Fig. 1). Consistent with previous classification efforts2, Cas13 subtypes cluster into four clades 13a–d. We found Cas13b to be most widespread, yet predominantly found within Bacteroidota. In contrast, Cas13c and Cas13d subtypes appeared least common, primarily found in Fusobacteriota and Bacillota (formerly Firmicutes), respectively. We found Cas13a to be phylogenetically more widely dispersed, although relatively limited in total number of homologues, spread across Pseudomonadota (previously Proteobacteria), Bacillota, Bacteroidota and Fusobacteriota.
Fig. 1: Maximum-likelihood phylogeny of Cas13 proteins and their distribution across the bacterial tree of life.
The four known subtypes, Cas13a–d, each form their clade (inner track) with a skewed distribution across bacterial taxa (outer track). A Vibrio cholerae Cas9 (UIO88932.1) was used as the outgroup. Cas13 subtypes and microbial taxa that encode Cas13 are denoted in the colour bar.
Our results are consistent with previous CRISPR search endeavours, suggesting that Cas13 effectors are some of the rarest Cas proteins currently identified2. Although RNA-targeting type-III CRISPR-Cas systems are relatively abundant in bacterial phyla2, we wondered whether the sparse occurrence of Cas13 effectors means that generalized resistance (for example, through RNA recycling41) or specialized resistance (for example, through anti-CRISPR33) to Cas13 is relatively rare as well.
LbuCas13a is a potent anti-phage effector against phage T4Two parsimonious explanations for the phylogenetic distribution of Cas13 effectors are that either Cas13 effectors are relatively ineffective anti-phage systems, limiting their phylogenetic spread owing to evolutionary pressure, or that Cas13 effectors are potent anti-phage systems, but the fitness cost of their abortive-infection-like effects30,34 causes selection against cas13 loci. To explore these possibilities, we tested the anti-phage activity of the most- and least-widely dispersed Cas13 effectors on the basis of our analysis of bacterial phylogeny—Cas13a and Cas13d, respectively (Fig. 1). We selected LbuCas13a from Leptotrichia buccalis and RfxCas13d from Ruminococcus flavefaciens due to their extensive biochemical characterization29,42,43,44,45. We additionally selected an engineered variant of LbuCas13a (eLbuCas13a) that was recently reported to have lower basal trans-RNA cleavage activity and, thus, reduced toxicity when expressed in E. coli45. Notably, none of the Cas13 orthologues here have been investigated for anti-phage activity. While a Cas13a orthologue from Listeria seeligeri has been used to restrict temperate and nucleus-forming phages19,30,33,35, LbuCas13a comes from a phylogenetically distinct sub-clade of Cas13a effectors (Supplementary Fig. 1).
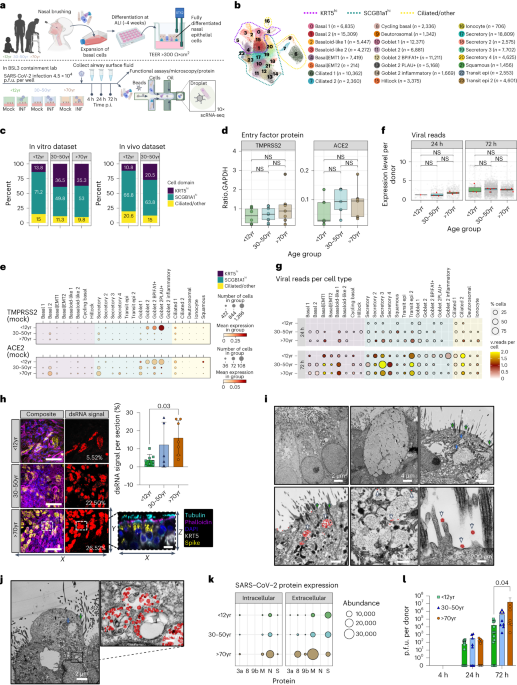
To establish an E. coli phage challenge assay for LbuCas13a and RfxCas13d, we created ‘all-in-one’ plasmids for inducible expression of cas13 using anhydrotetracycline (aTc) alongside a constitutively expressed crRNA (direct repeat-spacer) (Fig. 2a,b). During phage infection, phage RNAs are transcribed, including a crRNA-targeted transcript (orange, Fig. 2a). Upon recognition, Cas13 activates HEPN-mediated RNA cleavage, although the extent of trans-cleavage may be reduced for Cas13d relative to Cas13a43. Depending on the extent of Cas13-mediated RNA cleavage, phage-encoded Cas13 resistance, protospacer mutation rate and phage-encoded function containing the protospacer, phage may overcome the resulting general transcript degradation.
Fig. 2: Comparison of Cas13a and Cas13d in E. coli phage challenge assays with lytic phage T4.
a, Experimental architecture of Cas13 phage defence. Cas13 is expressed under aTc control alongside a crRNA. During phage infection, Cas13 unleashes toxic cis- and trans-cleavage if Cas13 detects its crRNA target. b, crRNA architecture employed in this study. c, Overview of T4 genes and transcript locations targeted by Cas13 in T4 phage challenge experiments. Approximate gene architecture is shown in forward orientation. crRNA locations are highlighted in orange. d, T4 phage infection in bacteria expressing phage-targeting crRNA and either LbuCas13a or RfxCas13d. EOP values represent the average of three biological replicates for a single crRNA. EOP data are presented as mean ± s.d. e, T4 phage plaque assays comparing the efficacy of Cas13a and toxicity of Cas13d. A representative plaque assay from three biological replicates is shown. An RFP-targeting crRNA is shown as a negative control.
To test the phage-restriction capacity of LbuCas13a and RfxCas13d outside their native context, we individually targeted a small panel of genes in phage T4. Phage T4 is a classical virulent dsDNA phage with a 169 kb genome and well-characterized genetic content46,47. From the perspective of phage genome editing, T4 represents an empirical challenge, displaying considerable variability in Cas-restriction efficacy for Cas9 and Cas12a, owing in part to modified glucosyl-5-hydroxymethylcytosine nucleotides26,27,36 and endogenous DNA-repair mechanisms20. For these reasons, we hypothesized that RNA targeting could be a superior strategy to inhibit T4 and related phages.
We designed a panel of Cas13 crRNAs targeting T4 transcripts with diverse design criteria (Fig. 2c)46. Targeted regions of T446 RNA sequences included essential genes (major capsid protein (mcp), transcriptional activator motA), a conditionally essential gene (deoxycytidylate hydroxymethylase gp42), a non-essential gene (accessory capsid protein soc), an early-infection gene (motA), a middle-infection gene (gp42), late-infection genes (mcp, soc), encompassing regions early in coding sequences (CDSs) (mcp, soc), middle in CDS (soc) and untranslated regions around the ribosome binding site (RBS) (gp42, motA) (Supplementary Table 1 and Fig. 2). We included a red-fluorescent protein (RFP)-targeting crRNA as a negative control. Broadly, this panel of crRNAs represents a careful exploration of Cas13 targeting the diversity of feature types present in a phage transcriptome.
Remarkably, in phage infection experiments, we observed robust phage restriction for all crRNAs tested using LbuCas13a (Fig. 2d). Independent of gene essentiality, timing of expression or position on transcript, we found that crRNA-guided LbuCas13a could restrict phage T4 over 100,000× when targeting mcp, gp42, motA or soc (Supplementary Fig. 2). In contrast, crRNA-guided RfxCas13d exhibited highly variable and less-efficient phage restriction. Further, RfxCas13d exhibited phage-independent E. coli growth inhibition during RfxCas13d expression (Supplementary Figs. 2–4), and we also observed a high degree of phage escape for RfxCas13d relative to LbuCas13a (Fig. 2e and Supplementary Fig. 2). It is possible that RfxCas13d lacks crucial components required for full phage defence or reduced toxicity, such as the WYL domain-containing proteins that appear in its native gene neighbourhood (Supplementary Fig. 5). Our results suggest that LbuCas13a is a remarkably potent single-protein defence system of phage T4 relative to other CRISPR-Cas systems20,26,27,36.
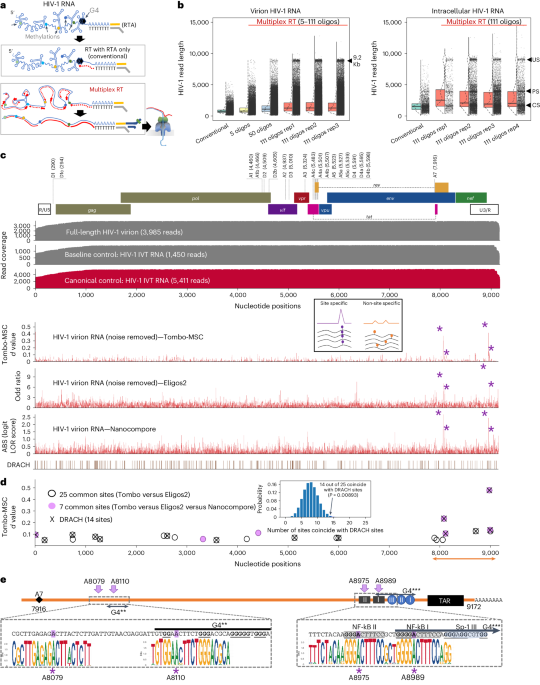
Cas13a confers resistance to diverse E. coli phagesTo the best of our knowledge, no single Cas effector (or antiviral defence protein) has been shown to confer broad-spectrum phage resistance against diverse dsDNA phages. To uncover the phage phylogenetic limits of Cas13a anti-phage activity, we challenged E. coli expressing LbuCas13a with a phylogenetically diverse panel of dsDNA E. coli phages. To generate a representative sampling of E. coli phages, we constructed a protein-sharing network from 2,307 phage genomes visualizing the relatedness of currently known E. coli phages (Fig. 3a). From this network, we assembled a panel of eight dsDNA and one single-stranded DNA (ssDNA) E. coli phages scattered across the E. coli phage phylogeny (Fig. 3a, Supplementary Figs. 6 and 7 and Table 2). This panel includes both model E. coli phages (T4, T5, T7, λ and M13) and non-model E. coli phages (EdH4, MM02, N4 and SUSP1). With the sole exception of phages T4 and MM02, these phages bear minimal nucleotide sequence similarity to each other (Fig. 3a and Supplementary Fig. 7). Furthermore, these phages have diverse lifestyles and reflect a realistic model sampling of the genetic diversity found among known E. coli phages. One of these phages displays temperate (λ), another displays chronic infection (M1348), while the remaining seven display obligately lytic life cycles. They comprise diverse lifestyles including documented plasmid-transfer-promoting (that is, ‘superspreader’)49, DNA compartmentalization16 and pseudolysogeny50 phenotypes. In aggregate, these phages not only represent genotypic diversity but also encompass a mixture of host-takeover strategies, modes of entry and degrees of previous characterization.
Fig. 3: Comparison of LbuCas13a anti-phage activity across dsDNA E. coli phage phylogeny.
a, Network graph representation of E. coli phages and their relatives. Nodes represent phage genomes that are connected by edges if they share significant similarity as determined by vContact276 (protein similarity). Nodes are shaded red if they are classified as an E. coli phage and blue if they only share similarity. Nodes are shaded black if they were assessed for sensitivity to LbuCas13a. b, EOP experiments for Cas13a designed to target an early or late transcript. EOP values represent the average of three biological replicates for a single crRNA compared to an RFP-targeting negative control crRNA. Phages T4, EdH4, λ, T5 and T7 have additional crRNAs that were tested and are presented in Supplementary Figs. 2, 8, 10, 14 and 15, respectively.
For each phage, we designed a pair of Cas13a crRNAs targeting either a putative early gene (DNA polymerase (dnap)), RNA polymerase (rnap (T7)), a lytic regulator (cro), replication protein (II (rep) (M13)) or a putative late gene (major capsid protein (mcp VIII) (M13)). An overview of Cas13-mediated phage restriction can be found in Supplementary Table 2, diversity of crRNAs tested in Supplementary Fig. 6 and a by-phage summary of results in Supplementary Figs. 2, 8–15. In aggregate, we observed substantial anti-phage activity for all 18 guides across the nine phages tested (Fig. 3b and Supplementary Table 2). Most crRNAs reduced phage infectivity 105–106-fold, with the sparse observation of mature plaque-forming units (p.f.u.). Across this entire study, we observed no mature p.f.u.s above 0.01% frequency in wildtype (wt) phage lysates (Supplementary Figs. 2, 8–15). We observed a single guide (targeting T5 dnap) to yield general toxicity and growth inhibition during LbuCas13a induction (Supplementary Fig. 16). This constraint required us to perform assays in the absence of induction, achieving a mere 102-fold restriction (Supplementary Fig. 14). However, employing the reduced-toxicity LbuCas13a mutant, eLbuCas13a45, we observed both phage restriction at 106-fold (Supplementary Fig. 14) and slightly reduced toxicity in the absence of phage (Supplementary Fig. 16). Thus, we believe that the subpar phage restriction by LbuCas13a was attributed to elevated background toxicity of the T5pol spacer rather than an inability to target this phage gene.
Interestingly, SUSP1 and M13 consistently displayed a small degree of resistance to Cas13a (Fig. 3b). Both early- and late- transcript targeting guides only decreased phage infectivity 5,000–10,000-fold compared with all other phages showing 105–106-fold infectivity reduction. We further investigated the efficacy of SUSP1-targeting crRNAs in a plate-reader assay at a wide range of multiplicities of infection (MOIs) (Supplementary Fig. 17). Compared to a non-targeting crRNA control, we found that both SUSP1dnap- and SUSP1mcp-targeting guides conferred phage resistance at all MOIs tested, including MOIs >10. These results indicate that Cas13a targeting not only conferred substantial population-level protection against SUSP1 infection, but also single-cell protection30. Potentially, this discordance with the abortive-infection model of Cas13 protection observed previously30 reflects a feature of LbuCas13a, a feature of fitness in a non-native host for Cas13 or a feature of SUSP1 and should be investigated further. Overall, we find that LbuCas13a is capable of anti-phage activity with no identified limits across the tested coliphage phylogeny.
A generalizable markerless method for editing phage genomesThe editing of virulent phage genomes has remained a major challenge for phage engineering and reverse genetics, largely due to the lack of universally applicable genetic tools or reliance on a native CRISPR-Cas system25,26,28,37,51,52,53,54. While the introduction of foreign gene content into phages is relatively straightforward to perform with homologous recombination (HR), ultimately the selection or screening for these rare recombinants is limiting even in well-characterized phages53. Given that LbuCas13a phage-restriction efficacy appears to have very little variability in terms of guide (Fig. 2), target (Figs. 2 and 3) and phage choice (Fig. 3), we suspected that Cas13a-mediated phage restriction would be an ideal tool for counterselection during phage genome editing. The high counterselection stringency observed earlier in this study obviates the need for selection markers, creating opportunities for multi-loci editing. Furthermore, the absence of protospacer-adjacent motif (PAM) requirements for LbuCas13a targeting29 suggests that virtually any position within or nearby a phage transcript could be edited and selected through LbuCas13a counterselection.
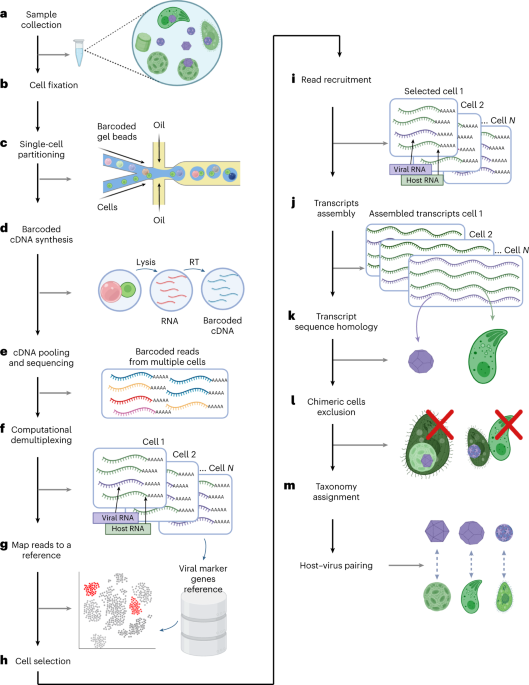
In principle, edits in the phage genome introduced through homologous recombination can escape LbuCas13a targeting, while wildtype phage cannot (Fig. 4a). To introduce and select for edits, we performed a simple two-stage homologous recombination and enrichment process (Fig. 4a, Supplementary Fig. 18 and Methods). Briefly, we employed two strains per edit: an editing strain containing a homologous recombination vector hosting a verification-primer binding site as well as 250 bp flanking phage homology arms, and a counterselection strain containing LbuCas13a and crRNA targeting the transcript carrying the locus to be edited. A pair of locus-specific examples are shown in Fig. 4b,c. We first infected the editing strain with wildtype phage at low MOI and collected the lysate consisting of a mixture of wildtype and edited phages (‘HR’ phage lysate) (Fig. 4a and Supplementary Fig. 18a). Then we diluted this lysate, infected the counterselection strain at low MOI and collected the resultant lysate (‘HR+E’ phage lysate) (Fig. 4a and Supplementary Fig. 18b).
Fig. 4: Cas13 facilitates a robust engineering strategy across diverse phages.
a, Overview of a simple two-step editing process. Wildtype phage T4 infects homology vector-containing strain at a low MOI, yielding a mixed population of wt (orange) and edited (purple) phages (‘HR’). This population is diluted and infects a LbuCas13a-expressing strain targeting the wt locus, enriching for edited phages relative to wt (‘HR+E’). b, Example gene deletion design for T4∆soc. Top: gene organization of wt T4soc locus shown with approximate locations of soc protospacers (orange) and homology arms (pink box). Bottom: gene organization of edited T4∆soc locus. The encoded deletion removes both soc protospacers, enabling enrichment of edited phages. c, Example large multi-gene deletion design from T4gp52.1 to T4rIIB (T4wtGT7). Top: gene organization of wt T4GT7 locus shown with approximate locations of T4ndd and T4denB protospacers (orange) and homology arms (pink box). Bottom: gene organization of edited T4GT7 locus. The encoded deletion removes both soc protospacers, enabling enrichment of edited phages. d, Editing penetrance (Methods) from three engineering replicates of the editing and enrichment process shown in a for T4∆soc, T4GT7, T7∆gp1.7, EdH4∆gp004 and EdH4∆gp214. In all cases, ‘negative control crRNA’ refers to an RFP-targeting crRNA, ‘positive control crRNA’ refers to the corresponding phage’s mcp-targeting crRNA, ‘enrichment crRNA’ refers to the crRNA used during the enrichment step shown in a and ‘verification crRNA’ refers to the deletion-targeting crRNA not used during enrichment. The ‘verification crRNA’ for EdH4 yielded a very toxic phenotype to establish a titre and is denoted with a red asterisk. Editing penetrance data are presented as mean ± s.d.
As a proof of concept that such an editing approach is immediately applicable to reverse genetics in a diversity of phages, we designed a small panel of edits across phages T4, T7 and EdH4. In particular, we designed four single deletions (for example, Fig. 4b) encoding for edited phages T4∆soc, T7∆gp1.7, EdH4∆gp004 and EdH4∆gp214. While T4soc and T7gp1.7 (a nucleotide kinase) are known non-essential genes46,55 under standard laboratory conditions, phage EdH4 has neither been edited previously, nor is there any knowledge of its genes’ essentialities before this study. Thus, EdH4 represents a pressure test for how extensible this editing strategy is to other non-model phages. As an example of more complex edits, we also designed a large edit originally identified during forward genetic screens on T4 mutant T4GT736 (hereafter, this edit in the wildtype T4 background is referred to as ‘T4wtGT7’). This edit consists of a large 3.2 kb deletion in T4, fully deleting 12 genes and truncating T4gp52.1 and T4rIIB (Fig. 4c).
We designed two crRNAs disrupted by the edited phage locus of interest as well as an additional verification guide to confirm the entire gene deletion (examples for T4soc and T4wtGT7 are shown in Fig. 4b,c, respectively). When tested against wildtype phages T4, T7 and EdH4, candidate crRNAs for T4soc, T4ndd (nucleoid disruption protein), T4denB (endonuclease IV), T7gp1.7, EdH4gp004 (hypothetical protein) and EdH4gp214 (hypothetical protein) were approximately as effective in phage restriction as crRNAs targeting definitively essential genes such as mcp (Supplementary Figs. 2, 8 and 15). However, when targeting these putatively non-essential genes, we observed that plaques emerge at 10−3–10−4% frequency, potentially reflecting a low rate of mutative escape permitted by the genes’ non-essentiality. In line with the model of Cas13a primarily imparting phage defence through RNA trans-cleavage activity, these results indicate that the primary counterselection pressure does not depend on the essentiality of the crRNA target. One of these crRNAs, the verification crRNA for the EdH4gp004 deletion, displayed elevated toxicity upon expression and is the only crRNA in this study we could not get to function. Ostensibly, LbuCas13a’s auto-toxicity is due to the extensive self-complementarity within the spacer of the mature crRNA (Supplementary Fig. 19) and potentially reveals a design constraint to be explored in future studies.
After each stage of editing (Fig. 4a), lysates were collected and titred against counterselection strains expressing LbuCas13a targeting the wildtype version of the edited locus (‘enrichment crRNA’ and ‘verification crRNA’), targeting an unedited locus (‘positive control crRNA’ (mcp crRNA)) and targeting a non-existent locus (‘negative control crRNA’ (RFP crRNA)) (Supplementary Figs. 20–24). By comparing the estimated titre against the non-targeting crRNA, we obtained a phenotypic estimate of the relative prevalence of edits within the population (that is, editing penetrance). Before enrichment (‘HR’), we observed targeted Cas13a-resistant infectious centres at 0.01–1% frequency for all five edits (Fig. 4d). Of particular note, edits for EdH4 and T4wtGT7 were generally lower in abundance, suggesting lower HR frequencies of editing for EdH4, as well as larger modifications. Importantly, after enrichment (‘HR+E’), targeted Cas13a-resistant infectious centre edited phages comprised nearly 100% of the population, while emergent general Cas13 resistance remained low (Fig. 4d and Table 1). In addition to confirming edits using the verification crRNA, we also PCR-verified nine plaques from the ‘HR+E’ lysates spotted on the counterselection crRNA for each edit (Supplementary Figs. 25–29). Unbiased PCR-derived Sanger sequencing further confirmed that the nature of Cas13 resistance was due to the designed edit in all cases.
Table 1 Summary of Cas13a-mediated phage genome editingPAMless Cas13a enables minimal edits in phage genomesWe aimed to further take advantage of the flexibility enabled by Cas13a’s PAMless nature by creating and enriching minimal edits that only Cas13a could easily select for20,26,27,36, using T4 as a model virulent phage. We designed six mutants at either the non-essential soc gene or essential dnap using silent mutations, thus ‘recoding’ the target gene (Fig. 5). We designed these mutants to recode only a single codon (soc-C, dnap-C), recode the entire seed region (soc-S, dnap-S)
留言 (0)