
記住我
Surprisal analysis (SA) was applied on the single-cell level so that each cell could be plotted according to its molecular aberrations and network reorganization (Figs. 1 and 2).
Fig. 1
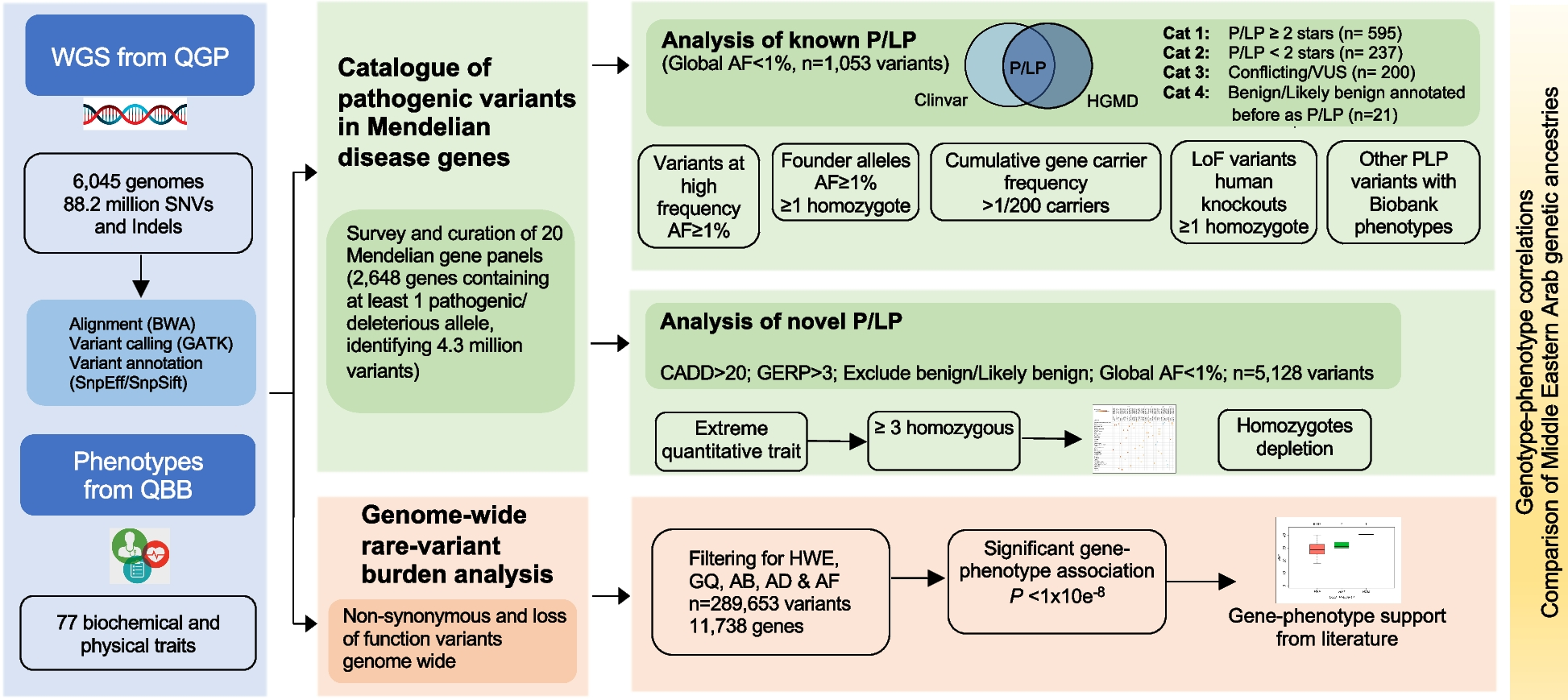
Study scheme. A A literature search was used to compose a list of oncomarkers for single-cell quantification (left panel). Selected cell-surface oncoproteins were quantified using multicolor FACS (right panel). B SA was extended to single cells to identify distinct subpopulations based on sets of unbalanced processes (cell-specific signaling signatures, CSSS, right panel). C To validate the hypothesis that targeting evolving cellular subpopulations in response to RT would enhance TNBC RT sensitization, a series of in vitro and in vivo experiments, using TNBC patient-derived and mice models, were performed
Fig. 2
Schematic of the application of the surprisal analysis algorithm. A Preparation of fluorescently-tagged single-cell suspensions from different sample sources (control and post-RT) for multicolor FACS analysis. Each cell was labeled with a mixture of 11 fluorescently tagged antibodies. B Surprisal analysis reveals protein expression level distributions at the reference (steady) state and the deviations thereof due to constraints in the system (e.g., irradiation). An example for calculated distribution of the expression levels at the reference state and deviations thereof is presented for Her2, initially quantified by FACS and analyzed by SA, in 4T1 mice model of TNBC. C Proteins deviating from the steady state in a coordinated manner are grouped into altered subnetworks referred to as “unbalanced processes.” For example, in one 4T1 cell, the levels of Her2 and EGFR deviate significantly (upregulated) from the steady state and in the other cells, cMet levels deviate significantly (upregulated as well). Thus, the two cells are defined by the analysis as possessing different processes. D The unbalanced processes in each cell provide a cell-specific signaling signature (CSSS). Each CSSS is schematically transformed into a cell-specific barcode, indicating active and inactive processes. E Cells sharing the same barcode are organized into distinct subpopulations. F Tumor-specific targeted therapy combinations are tailored against the subpopulations expanding in response to RT
Surprisal analysisSA is a thermodynamic-based information-theoretic approach [22,23,24] which has recently been implemented to analyze bulk [16, 19, 20, 25] and single-cell biological data [25, 26]. The analysis is based on the premise that biological systems reach a balanced state when the system is free of constraints [27,28,29]. However, when under the influence of environmental (e.g., exposure to a drug) and/or genomic constraints (genomic mutations that affect transcript/protein expression and function), the system is prevented from reaching a state of minimal free energy, and instead reaches a state of higher free energy—a constrained state [16].
Expression levels of various macromolecules, e.g., transcripts or proteins are used as an input for SA. Since the varying constraints that act upon living cells ultimately manifest as alterations in the cellular protein/gene expression network, they are viewed as emerging unbalanced molecular processes [19, 30]. Recent examples of SA implementation in biology include the characterization of bulk proteomic changes in large datasets, including multiple patient tissues and cancer cell lines, to predict a change in the behavior of systems [17, 18] or to design individualized drug therapies [19,20,21].
In this study, we analyzed protein expression data obtained from multicolor FACS in which each cell was labeled with a mixture of 11 fluorescently tagged antibodies. Additional file 1 provides the details for the models and single-cell analyses used in the study. It is important to note that this methodology may be applied to any single-cell proteomics data. The data matrix obtained from the flow cytometry analysis (~100,000–500,000 cells), in which columns are protein expression levels and rows are single cells, was used as an input for surprisal analysis. RT treatment imposes a constraint, but more than one constraint may be identified in the system.
Equation 1 was used to identify different constraints within tumor cells:
$$\underbrace_\mathrm\\\mathrm\;\mathrm\;\mathrm\;i\end}=\underbrace_\mathrm\;\mathrm\;\mathrm\;i\\\mathrm\;\mathrm\;\mathrm\;\mathrm\end}\exp\underbraceG_\lambda_\alpha(cell)\right)}_\mathrm\;\mathrm\;\mathrm\;\mathrm\\\mathrm\;\mathrm\;\mathrm\;\mathrm\;\alpha=1,2,..\end}$$
(1)
Here, \(_i^o(cell)\) is the expected expression level of protein i at the reference state in a measured cell. The exponential term in Eq. (1) represents the deviation from the reference value due to the constraints, including those imposed by irradiation. Giα are weights (the degree of participation) of a protein i in the unbalanced processes α = 1, 2... Proteins deviating in a similar manner from the steady state are grouped into unbalanced processes (see examples for 2 cells quantified in the 4T1 TNBC model in Fig. 2C). λα(cell) is the amplitude of an unbalanced process α = 1, 2. . in each tested cell. For example, Additional file 2: Fig. S2 presents λα(cell) values for process 8 (the network representing process 8 is shown in Fig. 3F and generated as explained below) in untreated and irradiated cells (6 days post-RT) in the 4T1 in-vitro model. Negative/positive amplitude denotes how the cells are correlated with respect to a particular process. The processes are indexed α = 1, 2, 3… so that the significance of the process decreases with an increasing index, i.e., unbalanced process 1 appears in a higher number of cells than unbalanced processes 2, 3, etc. Several unbalanced processes may be found in a system, however not all processes are active in all cells (see in the sections below how we define whether a particular process is active in a tested cell). ∑α = 1Giαλα(cell) represents the amount of information we have about each protein i. The partial deviations in the expression level of protein i due to the different constraints add up to the total change in expression level. A protein that is influenced by constraints, i.e., is influenced by one or more unbalanced processes, cannot take on any possible expression level. Its expression level is affected by the expression levels of other proteins in the unbalanced process in the cell.
Fig. 3
Resolving expanded 4T1 cellular subpopulations post-RT. A FACS expression levels of Her2 and cMet following RT. B–D Correlation plots between Her2 and cMet (B), Her2 and EGFR (C), and MUC1 and cMet (D) in irradiated cells. E Correlation plot between Her2 and EGFR levels expressed in the cells found to harbor process 3 (only cells with significant amplitudes (λ3(cell) values) were included in this plot, also see the “Methods” section). F Protein–protein networks were generated using single-cell SA analysis and STRING to assign the functional connections. To determine the direction of change in every protein (i.e., upregulation or downregulation) a sign of the amplitude in a process α in each cell was considered. Four unbalanced subnetworks (processes) out of 10 resolved in 4T1 (Additional file 3) are shown. G Each cell was assigned a barcode representing a cell-specific signature (CSSS). The most abundant (>1%) subpopulations are presented. H Based on these CSSSs the tumor was divided into distinct subpopulations. Quantification of subpopulations was performed using at least ~30,000 cells from each condition, which were obtained from at least three flasks and from at least three independent experiments for each time point. For A: statistically significant differences between control and 5 Gy; control and 15 Gy; and 5 Gy and 15 Gy were determined using a two-tailed Student’s t test (*P < 0.01)
Calculation of λ α(cell) and G iαWe fit the main Eq. (1), to the logarithm of the measured expression level of protein i in each cell using singular value decomposition (SVD). In practical terms, a matrix is constructed, containing the natural logarithm of protein expression levels in the different cells [26]. The procedure then utilizes SVD as an intermediate step, which calls for the construction of two square (and symmetric) co-variance matrices. One is smaller with a maximal rank of 11 (as the number of the proteins) and the second is bigger (depends on the number of cells, in this case at least 100,000). These matrices are diagonalized to calculate eigenvectors and eigenvalues. SVD and all other mathematical calculations described here were implemented using Matlab. Codes can be found in Github [31]. Eigenvectors and eigenvalues are further used by SA to calculate the amplitudes of the processes: λα(cell) for each cell if we use single-cell data, λα(k) for each sample k if we use bulk data [19, 20, 30], and Giα values, which are weights of the proteins in each process α. A detailed, step-by-step description of the mathematical procedure, namely how the eigenvectors and eigenvalues are used to calculate the amplitudes (λα(cell) or λα(k)) and Giα values, is described in the Supplementary Information of reference [16]). Any additional information regarding the mathematical procedures/codes can be provided upon request.
The number of calculated constraints is limited by the smaller dimension of the input matrix. In this case, it was limited to 11 (the number of measured proteins) and therefore a maximum of 10 constraints or unbalanced processes (10 constraints plus steady state) could be found. Calculations of the parameters using a smaller matrix (detailed in [16]) allow for the fast and efficient data processing of hundreds of thousands of cells. The number of proteins quantified in each cell can be significantly expanded (hundreds or thousands) without significantly increasing the data processing time.
Generation of protein-protein networks representing unbalanced processesAdditional file 2: Fig. S1 complements Fig. 3F and shows additional unbalanced processes active in the 4T1 system. The goal was to generate unbalanced processes composed of proteins with significant Giα values. Giα sign indicates the correlation or anti-correlation between proteins in the same process (Additional file 3). Upregulation or downregulation due to process α can be defined further using a product Giαλα(cell) for each protein in every cell in each experimental condition/time point: e.g., proteins with positive λα(cell) and positive Giα will be upregulated due to process α, since the product Giαλα(cell), which represents a deviation from the steady state due to process α, will be positive. Protein-protein interactions in each unbalanced process are based on the STRING database [32]. The radius of each circle in the map corresponds to the Giα value (Fig. 3F).
Calculation of cell-specific barcodes based on CSSSIt is important to note that not all processes are active in all cells. The term λα(cell) represents the importance of the unbalanced process α in the cell. Its sign indicates the correlation or anti-correlation between the same processes in different cells. To further map distinct subpopulations within the entire cellular population, we grouped cells sharing the same set of unbalanced processes, indicated by the cell-specific signaling signatures (CSSS), into distinct subpopulations (Figs. 1 and 2). Only unbalanced processes with significant amplitudes were included in the CSSS of each individual cell as follows:
To determine threshold limits for λα(cell) values, λα(cell) values were sorted and plotted as sigmoid plots in each process. Only λα(cell) values located on the tails of the sorted distributions were considered and used further for the barcode calculations (Additional file 2: Fig. S2).
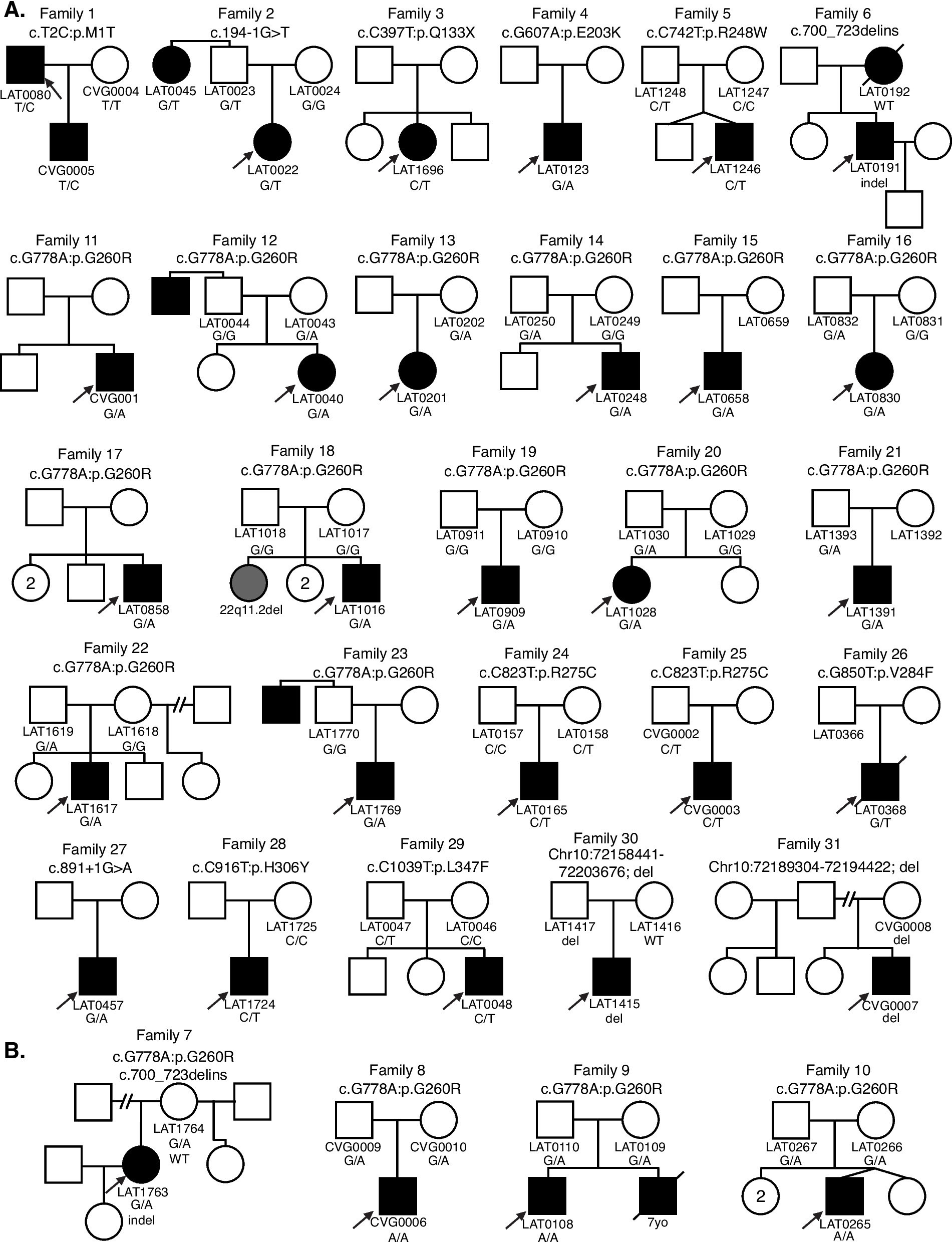
The combinations of unbalanced processes (CSSS) for each cell were generated using λα(cell) values that exceeded threshold limits. In this way, CSSSs were assigned to each cell and were converted into cell-specific barcodes for simple representation. Additional file 3-Additional file 8 include the input FACS data and the output parameters obtained using CSSS analysis (λα(cell) and Giα, and barcodes denoting subpopulations) for the major cancer systems, 4T1 and BR45, which have been tested in vitro and in vivo. Based on these cell-specific barcodes, distinct subpopulations were determined in the tumor (Fig. 3G, H). Subpopulations b and f (Fig. 4A) expanded significantly as detailed in the main text.
Fig. 4
Two distinct subpopulations expand and show proliferative properties in response to RT. A Very small subpopulations (<1%), represented by barcodes b and f, expanded significantly following RT (fold change in % of cells relative to the control of each time point). B–G 4T1 cells were irradiated with 15Gy. 6 days post-RT, cells were incubated with antibodies against Ki67, cMet, and Her2 and nuclei were stained with DAPI (fluorogel II). B, E 40× lens; scale bar represents 50 μm. C, F Sum intensities of Ki67 (C, left panel); Her2 (C, right panel); Ki67 (F, left panel); and cMet (F, right panel) were calculated from 8 to 10 fields using the NIS-Elements software (Nikon). D, G Correlation plots between D Ki67 and Her2 and G Ki67 and cMet were generated for each indicated condition to test co-activation represented in C, F. R values indicating the degree of correlation between Ki67 and Her2 (D) and Ki67 and cMet (G) were calculated before and after RT. H Survival rates of 4T1 cells in response to Trastuzumab (T), Crizotinib (C), RT, RT+C, RT+T, and RT+T+C as detected by MB survival assays 6 days post RT (upper panel), and cell viability as measured by the MTT assay (lower panel). Drugs were added from 3 days prior to RT until the end of the experiment. I Key downstream to Her2 and cMet signaling proteins are shown following different treatments. The predicted combination induced high levels of cleaved caspase-3 compared to radiation alone, irradiation+T, and irradiation+C. Downregulation of pAKT, pERK, and p-S6 was detected when T+C was applied prior to RT. For A: quantification of subpopulations was performed using at least ~30,000 cells from each condition, which were obtained from at least three flasks and from at least three independent experiments for each time point. For C and F, statistically significant differences between all presented conditions and the cells treated with RT+T+C were determined using a two-tailed Student’s t test (*P < 0.05); for H, upper and lower panels *P < 0.01
CSSS vs PCA and tSNESeveral dimensionality reduction algorithms have been developed to interpret single-cell variations (e.g., variations in protein or gene expression levels), such as clustering-based t-SNE analysis [33] or principal component analysis (PCA) [34,35,36]. These methods are very useful in the statistical determination of dominant expression patterns but are limited when a more deterministic partitioning of a tumor mass into cellular subpopulations, based on cell-specific sets of altered molecular processes, is required. For example, t-SNE is a non-deterministic method (e.g., different runs with the same hyperparameters may produce different results) and is unable to assign a certain protein to several processes, or to determine which processes are active in every cell (Additional file 2: Fig. S3). Therefore t-SNE will be less efficient when the determination of robust cell-specific signaling signatures is required (e.g., for drug combination design). Similarly, PCA focuses mainly on the most dominant patterns obtained from proteins with the highest variability in the population, rather than on cell-specific sets of altered processes [30, 37]. Additional file 2: Fig. S3 and Fig. S4 show separation of the 4T1 single-cell data, obtained using either t-SNE or PCA analysis (performed using Python), using 2 main principal components. Minority separation between control and RT-treated cells and within RT-treated cells can be observed and therefore CSSS analysis was vital in identifying the two separate subpopulations, b and f, that expanded in response to RT, and were mapped and quantified (Additional file 2: Fig. S3 and Fig. S4, see also Figs. 3-4 and main text for more details).
Experimental methodsPatient-derived tissue used to establish BR45 tumorsPatient-derived tumors were established from a female patient with triple-negative, invasive lobular breast cancer. The tissue was derived from the local chest wall recurrence, s\a mastectomy, chemotherapy, and radiotherapy. When implanted into the NSG mice the tissue formed a tumor, and then was used for the in-vivo experiments as described below.
Cell linesMurine 4T1 cells were kindly provided by Dr. Zvika Granot. MDA-MB-468 and MDA-MB-231 cells were acquired from ATCC and authenticated by the Genomic Center of the Technion Institute (Haifa). BR45 PDX were obtained from the Oncology Department at Hadassah –Jerusalem Medical Center with prior written informed consent. The BR45-derived and 4T1 cells were maintained and irradiated, after which flow cytometry was performed as indicated in Additional file 2: Supplementary Information file (SI).
Murine models Syngeneic model2.0×105 4T1 cells were inoculated subcutaneously into 6–7-week-old female BALB/c mice (ENVIGO).
Allogeneic modelBR45 tumors were induced in NSG (Jackson Laboratory) female mice either by injecting 4.0×106 cells or by transplanting xenografts, orthotopically.
After reaching the initial volume 80–100 mm3, mice were randomly grouped to approximately 8–10 animals per cage, and treatment was initialized. Tumor sizes were routinely measured with an electronic caliper every two days and their volumes were obtained using the formula V = (W (2) × L)/2. All in vivo experiments were performed with the approval of the Hebrew University of Jerusalem IACUC. See Additional file 2: SI Methods for more details.
In vivo treatmentsHigh dose rate (HDR) brachytherapy (GammaMed™ HDR, Iridium 192) was performed as previously described [38]. 12 Gy was administered on two alternative days. Trastuzumab was purchased from Teva Pharmaceutical Industries Ltd. Crizotinib (cMet inhibitor, #12087-50) and Erlotinib (EGFR inhibitor, #10483-1) were purchased from Cayman Chemical. (See Additional file 2: SI Methods for doses and regimens).
Cohort description for each type of in vivo experiment performed 4T1 In vivo experimentFirst experiment: 4T1 cells (2 ×105) were subcutaneously injected in female BALB/c mice.
Mice that reached a tumor volume of 80–100 mm3 were divided into two groups: control and irradiated. The irradiated group was treated by brachytherapy on two alternate days (12 Gy/day). Each group had two exit points: 6 and 12 days. Control samples n=12 (5 mice at day 6 post-RT and 7 mice at day 12 post-RT); RT-treated samples n= 17 (9 mice at day 6 post-RT and 8 mice at day 12 post-RT).
At the end of each time point mice were terminally anesthetized with Ketamine-Xylazine (150 mg/kg/20 mg/kg) IP, after which mice were euthanized by cervical dislocation. An incision was made and the tumor mass as a whole was gently separated from the conjunctive tissue using a sharp blade. Six and 12 days post-RT, whole tumors were collected for FACS analysis after mechanical cell dissociation. CSSS analysis was performed using FACS output.
Second experiment: The setup and procedure of collecting and analyzing the output was exactly as described in the first experiment. When tumors reached volumes of 80–100 mm3, mice were divided into four groups: each group had two exit points — 6 and 12 days. Mice were treated with 5 mg/kg Trastuzumab (T) and 25 mg/kg Crizotinib (C) starting 3 days prior to brachytherapy until the end of the experiment (day 17). Control: n=12 (7 mice at day 6 post-RT and 5 mice at day 12 post-RT), RT: n= 11 (5 mice at day 6 post-RT and 6 mice at day 12 post-RT), RT+T+C: n= 7 (4 mice at day 6 post-RT and 3 mice at day 12 post-RT), T+C: n= 10 (5 mice at day 6 post-RT and 5 mice at day 12 post-RT), RT+T: n= 12 (6 mice at day 6 post-RT and 6 mice at day 12 post-RT). For more details see Additional file 1.
BR45 PDX in vivo experimentsA small portion (~30 mm3) of BR45 PDX was transplanted orthotopically into each NSG female mouse. When tumor volumes reached 80–100 mm3, mice were divided into seven groups; each group had two exit points: 6 and 12 days. Mice were treated with 5 mg/kg Trastuzumab (T), 25 mg/kg Crizotinib (C), and 12.5 mg/kg Erlotinib (E) starting 3 days prior to RT until the end of the experiment (day 17). Control: n=6 (3 mice at day 6 post-RT and 3 mice at day 12 post-RT), RT: n= 5 (3 mice at day 6 post-RT and 3 mice at day 12 post-RT), RT+T: n= 6 (3 mice at day 6 post-RT and 3 mice at day 12 post-RT), RT+C: n= 6 (3 mice at day 6 post-RT and 3 mice at day 12 post-RT), RT+T+C: n=6 (3 mice at day 6 post-RT and 3 mice at day 12 post-RT), RT+T+C+E: n= 6 (3 mice at day 6 post-RT and 3 mice at day 12 post-RT), T+C: n= 7 (4 mice at day 6 post-RT and 3 mice at day 12 post-RT). Mice were irradiated by brachytherapy on two alternate days with two doses (12 Gy and 10 Gy). The setup procedure of collecting and analyzing the output data was exactly as described in the 4T1 model. For more details, see Additional file 1.
Flow cytometryEach sample was labeled with an 11 fluorescently tagged antibody (Ab) mixture. In addition when analyzing tumors, an exclusion cocktail including anti-mouse CD45, CD31 and CD140 was used in in-vivo experiments to exclude adjacent stromal and immune cells (Additional file 2: Table S8). A LSR-Fortessa Analyzer was utilized to measure all biomarkers simultaneously in each cell. See Additional file 2: SI Methods for more details.
Western blot analysisCell pellets were lysed with a 20% SDS buffer (targeted drugs were added 1 day prior to RT, which allowed to obtain enough protein concentration for WB). The protein content of each lysate was determined with a Pierce BCA Protein Assay Kit (#23225, ThermoFisher). Equal protein aliquots were subjected to SDS-PAGE (Criterion Stain Free, 4–15% acrylamide, Bio-Rad) under reducing conditions and proteins were transferred to a nitrocellulose membrane. (Millipore). Membranes were blocked with 5% non-fat milk for 1 hour at R.T. and probed with the appropriate antibody (Additional file 2: SI Methods), followed by horseradish peroxidase-conjugated secondary antibody (#123449, Jackson ImmunoResearch) and a chemiluminescent substrate (ECL #170-5061, Bio-Rad).
Survival assayCells were seeded at 70% confluency and treated as required for different time points. Cells were washed with PBS and fixed with 4% PFA for 10 min at R.T. The fixed cells were stained with methylene blue (MB) for 1 hour at R.T., washed and air dried overnight. The dye was extracted with 0.1M HCl for 1 hour at R.T. Absorbance was read at 630 nm.
MTT assayCells were seeded and treated as indicated in a 96-well plate for 6 days. Cell viability was determined using an MTT assay kit (#ab211091, Abcam). Equal volumes of MTT solution and culture media were added to each well and incubated for 3 h at 37 °C. MTT solvent was added to each well, and the plate was then covered with aluminum foil and put on the orbital shaker for 15 min. Absorbance was read at 590 nm after 1 h.
ImmunofluorescenceCells were grown on coverslips in six-well plates to reach 70% confluency by the next day, then fixed and permeabilized with cold absolute methanol. Afterwards, they were blocked with CAS blocker (cat. no. ZY-008120) and washed 3 times for 5 min with PBS, then stained with the following primary antibodies: Anti-mouse/human Ki-67 (BLG-151202), Rabbit Anti-Met (cMet) Polyclonal Antibody (BS-0668R), Neu (F-11) SC-7301. After washing 3 times with PBS for 5 min, cells were stained with secondary antibodies for 1 h at room temperature in the dark to visualize the aforementioned primary antibodies. The secondary antibodies conjugated to fluorophores were as follows: Goat anti-rat IgG H&L conjugated with Alexa Fluor 647 (1:400) (cat. no. 712605153), Goat anti-mouse IgG (H+L) conjugated with Alexa Fluor 488 (1:150) (cat. no. 115545003), and Goat anti-Rabbit IgG (H+ L) conjugated with Alexa Fluor 488 (1:150) (cat. no. 111545003). All secondary antibodies were purchased from Jackson ImmunoResearch. After washing 3 times with PBS, cell slides were mounted using fluorogel III mixed with DAPI (EMS, cat. no. 17985-01) to stain the nuclei. A spinning disk confocal microscope was used to visualize the expression of biomarkers of interest. The analysis was done using NIS-Elements software (Nikon).
Experimental statistical analysisSignificant differences between experimental conditions and experimental reproducibility were determined using the Student’s t-test (two tails, two samples equal variance); P values of ≤0.05 were considered statistically significant. All data was represented as the mean ± S.E. (standard error of the mean) if not indicated otherwise. Quantification of subpopulations was performed using a minimum of 30,000 cells from each condition, which were obtained from at least three flasks and from at least three independent experiments for each time point/condition. All experiments were performed minimally in biological triplicate if not indicated otherwise.
Code availability statementAll equations and mathematical procedures used in this article are detailed in the “Methods” section and/or referenced. The approach is covered by patent applications “A method for selecting patient specific therapy”, PCT/IL2019/050474, and “Methods of Determining Cancer Therapy,” PCT/IB2021/056136. Any additional clarification/information regarding mathematical procedures/codes can be provided upon request. The codes for single-cell computational analysis are publicly available from Github [31].
留言 (0)