
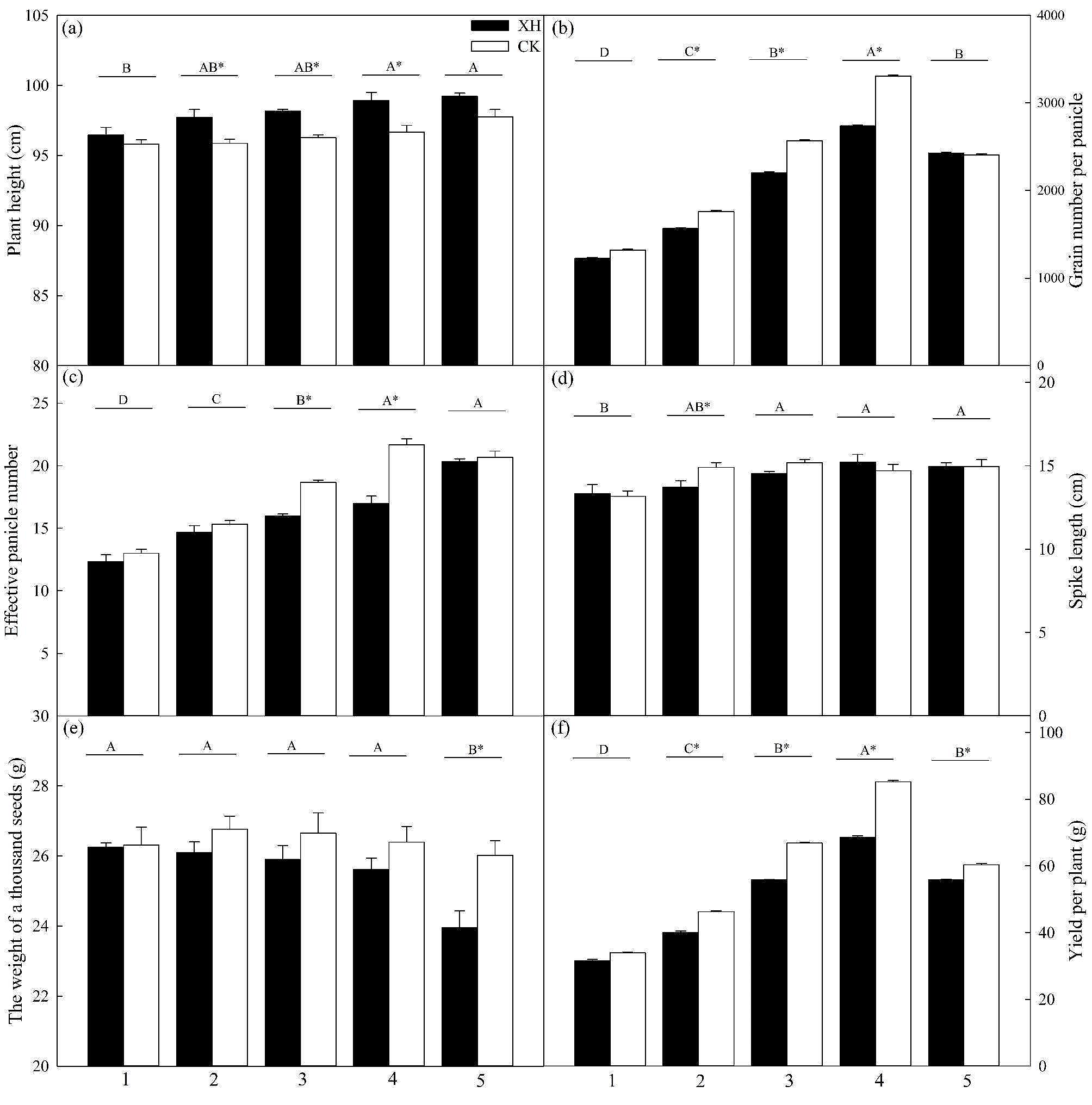
記住我





The housekeeping gene gyrA is considered to be more variable than 16S rRNA and has been used as a molecular tool for the classification and identification of B. subtilis species (Chun and Bae 2000; Borshchevskaya et al. 2013). In the genus Bacillus, the nucleotide diversity (Pi) of 16S rRNA and the gyrA gene sequences were 0.039 and 0.491, respectively (Fig. 1A, B blue line). It indicated that the degree of interspecies variation was significantly higher for the gyrA gene than for the 16S rRNA gene.
Fig. 1
Sliding window analysis of nucleotide variability [Pi (π)] along the sequence of the gyrA gene (A) and 16S rRNA (B) in Bacillus. 361 and 256 of gyrA and 16S sequences from 20 species and three species (B. amyloliquefaciens (n = 88), B. licheniformis (n = 71) and B. pumilus (n = 97)) were aligned with the Kalign method and nucleotide diversity was determined in DnaSP (v.6) using a window size of 100-bp, a step size of 10-bp, and points based on the mid-point of each window (i.e., the first point is at position 200). The blue lines display the Bacillus inter-species nucleotide diversity of the two genes and the orange, red and yellow lines display the nucleotide diversity of two genes in B. amyloliquefaciens, B. licheniformis and B. pumilus, respectively. The sequence length of the gyrA gene for analysis was 2450 bp, and that of 16S rRNA was 1540 bp
In three Bacillus species, the intraspecific nucleotide diversity (Pi) of 16S rRNA was again significantly lower than the intraspecific nucleotide diversity of gyrA gene sequences in all three species: B. amyloliquefaciens (Pi16S = 0.0014; PigyrA = 0.0244), B. licheniformis (Pi16S = 0.00024; PigyrA = 0.0021) and B. pumilus (Pi16S = 0.00136; PigyrA = 0.0344) (Fig. 1A, B nonblue lines).
In conclusion, the Bacillus gyrA gene shows higher variation rates than 16S rRNA, hence we propose that gyrA represents a promising molecular marker for analyses of Bacillus community diversity analyses and the diversity of Bacillus isolates.
First comparative tests of three primer pairs for the detection of Bacillus speciesAs indicated above Bacillus isolates have been already analyzed by primers targeting gyrA, however the specificity of these primers has not been investigated broadly (Roberts et al. 1994; Ansaldi et al. 2002). To satisfy the amplicon sequencing requirements, we designed a new primer pair (gyrA3) (Fig. 2A), and compared its amplification potential in colony PCR and virtual PCR with the previously designed primers, referred to here as gyrA1 and gyrA2 (Fig. 2A) (Roberts et al. 1994; Ansaldi et al. 2002).
Fig. 2
Displays the ability of the three primer pairs to amplify the gyrA gene for seven Bacillus strains. A The position of gyrA amplicons was obtained by primer pairs gyrA1, gyrA2 and gyrA3. B PCR amplification of seven Bacillus strains using three gyrA primer pairs. The orange square indicates that the target gene of gyrA was amplified by the indicated primer pair, and the white square indicates that it was not. The phylogenetic tree was constructed based on the complete 16S rRNA (1358 bp) (Additional file 2: Dataset S3) by using the Neighbor-Joining method in MEGA 5.05 software. The reliability of clades was tested by the 1000 bootstrap replications. C Display of the results of the computer simulation amplification ability of the three gyrA primer pairs using the gyrA sequence database. The white columns represent the number of sequences in the database that were matched by the primers, while the grey columns represent the number of Bacillus species identified by the primer pairs (Additional file 2: Table S6). The gyrA sequences in database were downloaded from the NCBI genome database (Additional file 2: Table S3)
First, we selected seven strains of different Bacillus species: L. fusiformis, P. polymyxa, B. pumilus, B. velezensis, B. megaterium, B. cereus and B. subtilis (Fig. 2B) to perform PCR amplification with primer pairs gyrA1, gyrA2 and gyrA3. The PCR amplification results showed that gyrA1 detected only B. subtilis; gyrA2 detected B. subtilis, B. velezensis and L. fusiformis; whereas gyrA3 performed much better and detected all Bacillus species included in the analysis (Fig. 2B and Additional file 1: Fig. S1).
The in-silico PCR analysis was performed using the gyrA gene database containing 226 Bacillus species. The results showed that only 8 Bacillus species were amplified in-silico by gyrA1, 9 Bacillus species were amplified by gyrA2 (Fig. 2C and Additional file 1: Fig. S2), while 55 Bacillus species were amplified by gyrA3 (Fig. 2C). The majority of sequences amplified by gyrA1 and gyrA2 belonged to B. subtilis, whereas gyrA3 demonstrated broader diversity as evidenced by the amplification of seven species and in-silico PCR (Fig. 2B, C and Additional file 2: Table S6).
Specificity range of the gyrA3 primer pair by using PCR and in silico PCRBecause the gyrA3 primer pair performed better than the previously reported primers, we next combined analysis of the in-silico amplified gyrA genes with PCR analysis of Bacillus isolates from our laboratory culture collection. Virtual gyrA3 PCR amplicons from 55 different Bacillus species from the gyrA gene database were evenly distributed among the branches of the phylogenetic tree (Fig. 3 orange and green).
Fig. 3
The amplification ability of the primer pair gyrA3 of Bacillus species and related species performed by computer simulation and in situ amplification. The Bacillus species indicated by the orange color (48 species) represent the species in the database that were matched by gyrA3 only in virtual PCR. The Bacillus species in blue color represented strains that can be amplified by gyrA3 only by colony PCR. These strains included 21 Bacillus species and 16 Bacillus’s related genera species. The Bacillus species in green color (7 species) represent strains that can be amplified by two methods: virtual PCR and colony PCR. The species in black color (2 species) represent strains that cannot be amplified in virtual PCR nor by colony PCR. The phylogenetic tree was constructed based on 16S rRNA (1302 bp)
Next, we used 127 strains of Bacillus (32 species) and related genera (Paenibacillus, Lysinibacillus, Aneurinibacillus, Virgibacillus, Brevibacillus, Halobacillus, Fictibacillus, 24 species) from our laboratory culture collection to amplify their gyrA genes with a gyrA3 primer pair (Additional file 2: Table S1). The results showed that 28 Bacillus species and 16 Bacillus-related species could be amplified by the gyrA3 primers (Fig. 3 blue and green), while the remaining 4 Bacillus species and 8 Bacillus-related species could not be amplified by the gyrA3 primers. Of these Bacillus species, marked in green in the phylogenetic tree, 7 species were detectable by both methods (Fig. 3 green). In summary, the primer pair gyrA3 can potentially detect 76 Bacillus species and as many as 16 species from related genera (Fig. 3).
The gyrA gene provides better intraspecific phylogenetic resolution than the 16S rRNA gene among certain speciesCompared to 16S rRNA, the molecular evolution rate of gyrA gene sequences is faster (Timmis and Ramos 2021), so we hypothesized that gyrA might provide better phylogenetic resolution at the subspecies level. The single nucleotide polymorphisms (SNPs) analysis of the 16S rRNA (V3-V4 region) and the gyrA gene (gyrA3 amplicon region) was performed in four Bacillus species (B. amyloliquefaciens, B. pumilus, B. megaterium and B. anthracis). We did not include an analysis of the B. subtilis genomes because templates for gyrA primers have already been developed and applied for analyses of this species (Roberts et al. 1994; Ansaldi et al. 2002; De Clerck et al. 2004; Stefanic and Mandic-Mulec 2009).
In B. amyloliquefaciens, the 480 bp long 16S rRNA region (V3-V4 region) contained 12 variable base sites (Fig. 4A and Additional file 1: Fig. S3A), which were detected in only 4 of 116 genomes of this species (Fig. 4A), and the SNPs frequency at variable sites within the 4 genomes ranged from 0.21%-1.46% (Additional file 1: Fig. S3C red column). In contrast, the 500-nucleotide gyrA region (positions 350–850) contained 59 variable sites (Fig. 4B and Additional file 1: Fig. S3B). The variable sites were detected in 109 of 116 genomes (Fig. 4B), and the frequency of SNPs at the variable sites ranged from 0.4% to 6.4% (Additional file 1: Fig. S3C blue column).
Fig. 4
Alignment of the 16S rRNA and gyrA sequences of B. amyloliquefaciens (116 genomes) and B. pumilus (140 genomes). Sequences were aligned using L-INS-I method of MAFFT (v7.487). The analysis involved positions along 16S rRNA from 330–810 and along gyrA from 350–850. On the left side of the graph GCF reference numbers of genomes were displayed, with reference sequence GCF_017815555.1 displayed at the top for B. amyloliquefaciens and GCF_014269965.1 for B. pumilus. The display of base site variation was drawn using MEGA (v.5.05). The colored line (red or blue) indicates a variation of specific base sites as compared to the reference sequence
In B. pumilus, alignment of the 16S rRNA V3-V4 region revealed 21 variable base sites (Fig. 4C and Additional file 1: Fig. S4A), but again only in 11 out of 140 genomes (Fig. 4C). The frequency of SNPs at variable sites ranged from 0.21% to 3.54% (Additional file 1: Fig. S4C red column). In contrast, in the gyrA gene, 130 of the base sites were variable (Fig. 4D and Additional file 1: Fig. S4B) and these were found in 87 of 140 genomes, with SNPs frequencies at variable sites ranging from 0.2% to 15.8% (Additional file 1: Fig. S4C blue column). However, in B. pumilus nearly 50% of the genomes examined had 100% identity in the gyrA gene, indicating a high degree of relatedness between genomes that may require sequencing of additional marker genes for clonality verification and strain typing.
We also observed that the variation of the gyrA gene is quite different in different Bacillus species, which could be a bias of the NCBI database or property of certain species. For example, B. megaterium showed lower diversity with 3 bases of variation in the 16S rRNA alignment region (V3-V4 region) (Additional file 1: Fig. S5A and Fig. S6A). Although 48 of 117 genomes showed polymorphism, the maximum SNPs frequency at variable sites of B. megaterium genomes was only 0.42% (Additional file 1: Fig. S6C red column). The gyrA gene was again more polymorphic with 58 bases of variation (Additional file 1: Fig. S5B, S6B) occurring in 99 of 117 genomes, with SNPs’ frequencies at variable sites ranging from 0.2% to 2.8% (Additional file 1: Fig. S6C blue column).
The variation divergence between the 2 genes of B. anthracis was much lower than in the three Bacillus species described above. Although we identified 13 variable base sites in the 16S rRNA V3-V4 region and 65 variable base sites in the gyrA gene region (Additional file 1: Fig. S5C, D and Additional file 1: Fig. S7A, B), the SNPs occurred in only 7 and 18 of 226 genomes, respectively. Moreover, the SNPs frequencies at variable sites in 7 and 18 of B. anthracis genomes were also low: 0.21%-1.04% and 0.2%-7.4%, respectively (Additional file 1: Fig. S7C).
Overall, our results showed that within Bacillus species the frequency of SNPs in the gyrA gene was consistently much higher than in the 16S rRNA (Fig. 4 and Additional file 1: Fig. S5). We therefore suggest that the gyrA gene provides better resolution than 16S rRNA for identification and typing of Bacillus isolates at the subspecies level. This is particularly true for B. amyloliquefaciens, B. pumilus and B. megaterium but less so for B. anthracis (Table 1).
Table 1 Polymorphisms of the 16S rRNA and gyrA geneThe resolution power of Bacillus mock community gyrA amplicon sequencingOur results above suggest that the amplicon sequence of the primer pair gyrA3 could be used as a molecular marker for diversity analysis of Bacillus. Next, we aimed to design a mock community to test the efficacy of the primers gyrA3 and used the general 16S rRNA primers (V3-V4) as a positive control. For better comparison, we designed 2 additional primer pairs, gyrA4 and gyrA5, which are very close to the position of the gyrA3 in the gyrA gene (Additional file 1: Fig. S8). We selected eight strains belonging to four species (B. altitudinis, B. licheniformis, B. velezensis and L. pakistanensis) and successfully amplified their gyrA gene by gyrA3, gyrA4 and gyrA5 primer pairs in a routine PCR for selected strains (Fig. 5A).
Fig. 5
The results of amplicon sequencing for the mock community including eight Bacillus strains. A PCR amplification of eight Bacillus strains by gyrA gene primer pairs. The orange square indicates positive and white square unsuccessful amplification. The amplicon sequencing results of 16S rRNA gene B and gyrA gene using primer pairs gyrA3 C, gyrA4 D, and gyrA5 E for the mock community. The relative abundance is represented by reads numbers for each unit as indicated on the X axis. The phylogenetic tree was contracted based on the complete gyrA genes (2469 bp) (Additional file 2: Dataset S2) by using the Neighbor-Joining method in MEGA 5.05 software. The reliability of clades was tested by the 1000 bootstrap replications. The box plots were drawn by the ggplot2 R package (v.3.2.1)
Next, we constructed a mock community of eight strains to retest the resolution power of the three gyrA primer pairs and the 16S rRNA-specific primers. Our goal was to test whether the primer pair is suitable for determining the diversity of the mock community (Fig. 5). Sequencing of the 16S rRNA amplicons showed that 16S rRNA primers could distinguish only five units, as strains LY1 and LY18, LY37 and LY43, and LY39 and LY48 had identical V3-V4 nucleotide sequence (Fig. 5B). The gyrA primer pairs were capable of resolving 6 units, but gyrA4 and gyrA5 produced amplicons of variable abundance and preferentially amplified LY35 and LY2, respectively (Fig. 5C–E). In comparison, primers gyrA3 also amplified six fragments but the relative abundance of these amplicons was more uniform (Additional file 2: Table S7) with the exception of LY2 strain (Fig. 5C). Our data suggest that gyrA3 has potential for Illumina amplicon sequencing of more complex Bacillus communities.
留言 (0)