Cell culture
The R28 retinal precursor cell line, an immortalized adherent retinal precursor cell line derived from infantile Sprague–Dawley rat retinas, is used for in vitro studies of neuroprotection, cytotoxicity, and physiological function of RGCs [10,11,12, 81]. The R28 cells were cordially provided by the Department of Anatomy and Neurobiology of Central South University (Changsha, China). The R28 cells were cultured in low-glucose DMEM (Gibco, Waltham, Massachusetts, USA) with 10% FBS (Cell-Box, China) and 1% penicillin/streptomycin (Gibco, Waltham, Massachusetts, USA). The cells were maintained in a humidified incubator at 37 ℃ and 5% CO2.
Animals
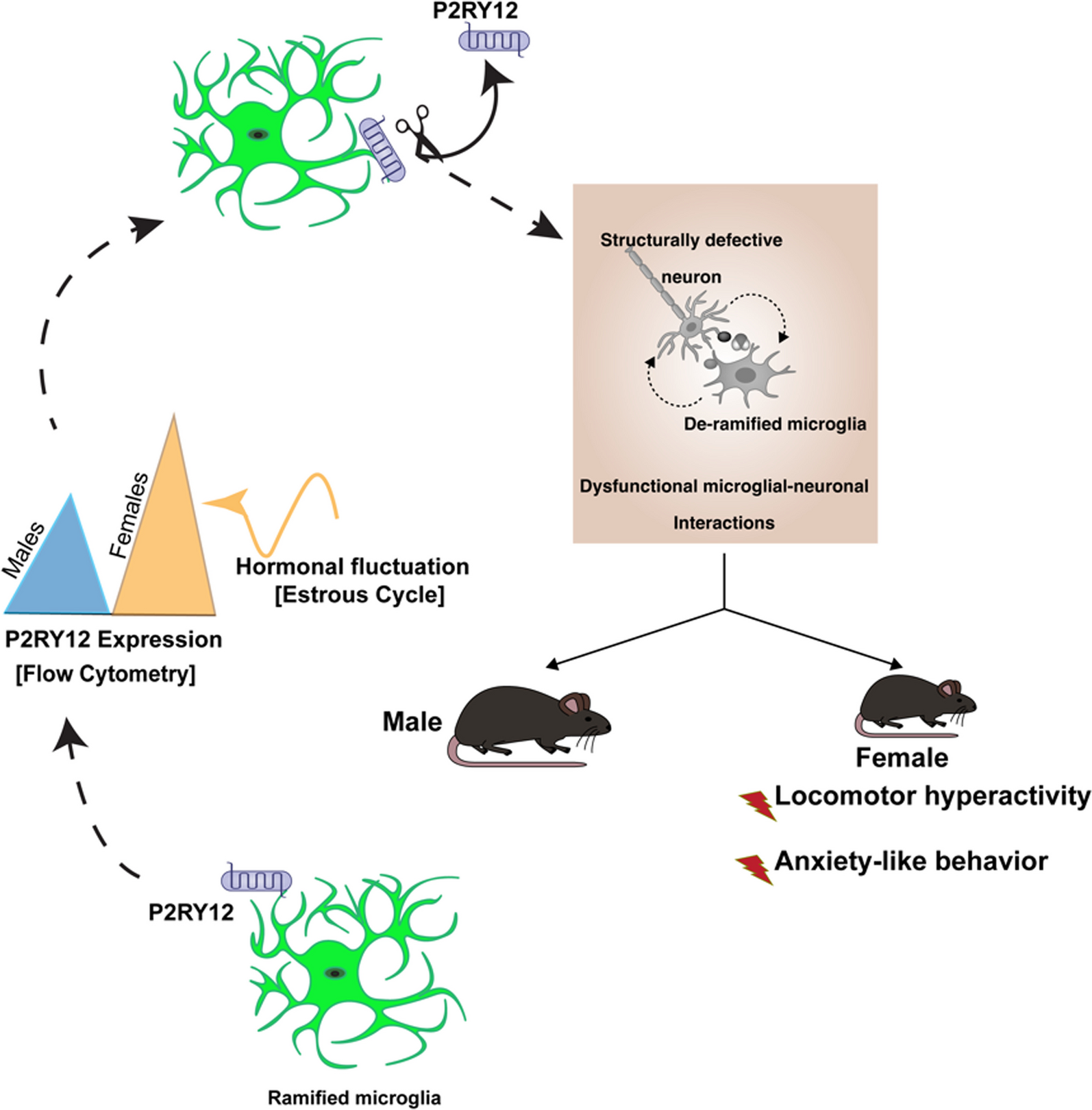
This study used 140 7-week-old (20–25 g) C57BL/6J mice. The animals were maintained under standard laboratory conditions of a 12-h cycle of light and dark at a temperature of 21 ± 1 ℃. Food and water were available ad libitum. All animals were treated according to the Association for Research in Vision and Ophthalmology Resolution on the Use of Animals in Research. The mice were randomly divided into seven groups of 20 mice each: saline group, NMDA 1 d group, NMDA 3 d group, NMDA 5 d group, NMDA + DMSO group, NMDA + GSK872 group, NMDA + Nec-1 group. The retinal excitotoxic damage model was achieved by intravitreal injection of 1 μL of 20 mM NMDA in saline (APExBIO Technology, Houston, TX, USA). Mice were anesthetized with an intraperitoneal injection of 1% pentobarbital sodium (10 mL/kg), followed by oxybuprocaine hydrochloride eye drops for topical anesthesia. Pupils were dilated with tropicamide eye drops. Subsequently, 1 μL NMDA was injected into the vitreous cavity of both eyes using a 33-gauge needle connected to a 10-µL syringe (Hamilton Company, USA). The control group received intravitreal injection with the same volume of saline. Antibiotic ophthalmic ointment was applied to the eyes post-injection. The animals were killed at 1, 3, and 5 days after intravitreal injection, and the eyes were enucleated and processed for further analysis.
Drug administration
For the in vitro analysis, R28 cells were seeded in 6-well plates at a density of 3 × 105 cells/well and incubated overnight before treatment with different concentrations of glutamate (Abcam, Cambridge, UK) and for different durations. GSK872 (Selleck, Houston, TX, USA) and Nec-1 (Sigma-Aldrich, St. Louis, MO, USA) were dissolved in DMSO and diluted in culture medium. The cells were divided into five groups according to different treatments: Control group (without treatment), Glutamate-treated, Glutamate + DMSO treated, Glutamate + GSK872 treated, and Glutamate + Nec-1 treated groups. Then the cells were incubated for 24 h before being analyzed.
For the in vivo analysis, mice received an intravitreal injection of 1 μL solution containing NMDA and GSK872 (80 μM in saline) or NMDA and Nec-1 (40 μM in saline). The group treated with the same volume of solution containing NMDA and DMSO was used as control. All treatments were given intravitreally in both eyes. Five days after injection, eyes were enucleated and processed for further analysis.
Observation of cell morphology
The R28 cells were seeded in 6-well plates at a density of 3 × 105 cells/well and incubated overnight. Then the cells were exposed to 10 mM glutamate for 6 h, 12 h, and 24 h. At each time point, cells were observed and photographed using phase-contrast microscopy (Nikon, Tokyo, Japan).
Cell viability assay
Cell viability was measured using a Cell Counting Kit-8 (CCK-8, NCM Biotech, China) according to the manufacturer’s instructions. The R28 cells were seeded at a density of 1 × 104 cells/well in 96-well plates and incubated overnight. Then the cells were treated with different concentrations of glutamate, GSK872, and Nec-1 for 24 h. Subsequently, 10 μL CCK-8 solution was added to each well and incubated for 2 h. The absorbance was measured at 450 nm using a spectrophotometer.
Lactate dehydrogenase release assay
An LDH release assay kit (Beyotime, Shanghai, China) was used to measure LDH released from necrotic cells after different treatments. In brief, cell cultures were collected from 96-well plates after being centrifuged at 400g for 5 min using a porous plate centrifuge and then the cell-free culture supernatants were incubated with the working reagent mixture in the dark for 30 min at room temperature according to the manufacturer’s instructions. Subsequently, the absorbance was measured with a spectrophotometer at a wavelength of 490 nm. The LDH release rate of R28 cells was calculated as the percentage of the absorbance value: LDH release rate% = (sample cells − control cells)/(LDH releasing reagent treated cells − control cells) × 100.
Hoechst 33342/PI dual staining assay
Hoechst 33342/PI dual staining (Beyotime, Shanghai, China) was used to measure the cell death rate of the R28 cells. After being treated with the target compounds, the R28 cells were incubated with cell staining buffer, Hoechst staining solution, and PI staining solution at 4 ℃ in the dark for 30 min according to the manufacturer’s instructions. After being washed with PBS, the cells were observed and photographed with a fluorescence microscope (Nikon, Tokyo, Japan). The photos were analyzed using ImageJ software. The percentage of cell death was calculated using the formula: cell death rate% = PI-positive cells/Hoechst-positive cells × 100. Five visual fields were selected in each sample.
Annexin V-FITC/PI double staining
Annexin V-FITC detection kit (Beyotime, Shanghai, China) was used to detect apoptosis and necrosis in R28 cells after glutamate treatment. After incubation with glutamate for different time points, R28 cells were collected using EDTA-free trypsin and then resuspended with PBS. After that, cells were stained with 5 μL of Annexin V-FITC and 10 μL of PI for 15 min at room temperature in the dark according to the manufacturer’s instructions. Next, the stained cells were analyzed by flow cytometry. The percentage of Annexin V+ cells indicated cell death rate, which was consisted of Annexin V+/PI− (early apoptosis) cells and Annexin V+/PI+ (necrosis and late apoptosis) cells while both Annexin V and PI negative cells were considered as living cells. For each sample, 3 × 104 cells were collected.
Transmission electron microscopy (TEM)
After being treated with the target compounds, R28 cells and retinas were harvested and then fixed in ice-cold 2.5% glutaraldehyde at 4 ℃ in the dark for 24 h. After being washed with PBS 3 times for 10 min, cells and retinas were fixed with 1% osmium tetroxide for 3 h, and then rinsed with PBS 3 times for 10 min. Next, the samples were gradient dehydrated with ethanol and acetone and embedded in epoxy resin followed by being cut into 50–60 nm sections. Then, the sections were observed under a HT7700 transmission electron microscope (HITACHI, Tokyo, Japan) after being stained with 3% uranyl acetate combined with lead citrate.
ROS production detection
The intracellular ROS level was measured using an ROS assay kit (Biosharp, China). In brief, after being treated with different compounds, the R28 cells were stained with 10 μM DCFH-DA at 37 ℃ in the dark for 30 min according to the manufacturer’s instructions. Then, the cells were observed and photographed under a fluorescence microscope (Nikon, Tokyo, Japan) after being washed three times with fresh medium. Moreover, the fluorescence intensity of DCFH-DA was measured by flow cytometry.
qRT-PCR
Total RNA was extracted from the R28 cells using TRIzol reagent (Invitrogen, Waltham, MA, USA), and cDNA was synthesized with a Hifair III 1st Strand cDNA Synthesis Kit (Yeasen Biotechnology, Shanghai, China). Quantitative real-time polymerase chain reaction (RT-PCR) was performed using a Hieff qPCR SYBR Green Master Mix (Low Rox, Yeasen Biotechnology, Shanghai, China) with a sequence detection system (Prism 7500, Applied Biosystems, Waltham, MA, USA) according to the manufacturer’s instructions. The specific primers were designed by Sangon Biotech (Shanghai, China), including TNF-α (forward 5′-ACCATGAGCACGGAAAGCAT-3′ and reverse 5′-AACTGATGAGAGGGAGCCCA-3′), IL-6 (forward 5′-TCTGGTCTTCTGGAGTTCCGT-3′ and reverse 5′-CTTGGTCCTTAGCCACTCCT-3′), IL-1β (forward 5′-TACCTATGTCTTGCCCGTGG-3′ and reverse 5′-TAGCAGGTCGTCATCATCCC-3′), and GAPDH (forward 5′-AGTGCCAGCCTCGTCTCATA-3′ and reverse 5′-TGAACTTGCCGTGGGTAGAG-3′). Relative mRNA levels were normalized to those of GAPDH and were calculated using the 2−ΔΔCT method. Each sample was measured in triplicate wells, and the experiments were repeated three times.
Western blot analysis
After being treated with the target compounds, proteins from the R28 cells and retinas were harvested using a RIPA lysis buffer (Beyotime, Shanghai, China) with Protease Inhibitor Cocktail (APExBIO Technology, Houston, TX, USA) and Phosphatase Inhibitor Cocktail (APExBIO Technology, Houston, TX, USA), and then the retinas were homogenized at 4 ℃ for 15 min. Next, the lysates were centrifuged at 12,000g and 4 ℃ for 20 min. Then, the supernatant was transferred into a fresh tube and subjected to sonication, clarified by centrifugation (12,000g, 4 ℃ for 10 min), and the supernatant was collected. The protein concentration was measured by bicinchoninic acid assay (Beyotime, Shanghai, China). After being mixed with 5 × loading buffer and boiled at 95 ℃ for 10 min, the protein was subjected to electrophoresis on an SDS/PAGE gel and transferred to PVDF membranes. After blocking with 5% skim milk for 90 min, the membranes were incubated overnight at 4 ℃ with the following primary antibodies: anti-RIP1 (#3493, 1:1000, Cell Signaling Technology, Danvers, MA, USA), anti-RIP3 (#15,828, 1:1000, Cell Signaling Technology, Danvers, MA, USA), anti-MLKL (ab243142, 1:2000, Abcam, Cambridge, UK), anti-MLKL(phospho S345) (ab196436, 1:1000, Abcam, Cambridge, UK), anti-NLRP3 (ab263899, 1:1000, Abcam, Cambridge, UK), anti-caspase-1 (22915-1-AP, 1:2000, Proteintech, Rosemont, IL, USA), anti-IL-1β (ab254360, 1:1000, Abcam, Cambridge, UK), anti-GAPDH (60004-1-Ig, 1:20000, Proteintech, Rosemont, IL, USA). Then, the membranes were washed three times with TBST for 10 min and incubated with the respective peroxidase-conjugated secondary antibodies (Zen Bio, Chengdu, China) for 2 h at room temperature. After three 10-min washes, the proteins were visualized using an enhanced chemiluminescence (ECL) method (NCM Biotech, China) with a chemiluminescence imager (Bio-Rad, California, USA) according to the manufacturer’s instructions. The integrated optical density values of specific proteins were quantified using ImageJ software. GAPDH was used as the internal control.
Measurement of glutathione (GSH)
GSH was detected using a GSH and GSSG Assay Kit (Beyotime, Shanghai, China). In brief, cells were collected after being washed with PBS and then mixed with the protein removal reagent (M) solution. Then, the samples were freeze–thaw twice using liquid nitrogen and 37° water bath followed by ice bath for 5 min. Next, several working solutions of the assay were prepared according to the manufacturer’s instructions. After incubation with the working solutions for the indicated times, GSH levels were measured by detecting the absorbance value of all samples at 412 nm using a spectrophotometer.
Hematoxylin and eosin (HE) staining
After the mice were killed, their eyes were enucleated at the designated time points, fixed with 4% paraformaldehyde (PFA), and embedded in paraffin. Four paraffin-embedded retinal tissue sections of each eye were cut in a vertical meridian through the optic disk at 4 µm thickness and stained with HE. Micrographs of the stained retinal sections were captured using a light microscope (Leica, Wetzlar, Germany). Total retinal thickness (the internal to the outer limiting membrane) and GCC thickness (from the INL to the nerve fiber layer) were measured within the area of 1 mm distance to the optic nerve. Each measurement was performed four times and averaged.
Immunofluorescence staining of retinal whole-mounts
At different time points post intravitreal injection, the mice were killed and perfused with saline and 4% PFA. Eyes were then enucleated and fixed in 4% PFA for 2 h at room temperature. Subsequently, retinas were isolated from the eyecups, and each retina was divided into four quadrants. Next, retinas were incubated with blocking buffer containing 5% bovine serum albumin (BSA) and 0.5% Triton-X 100 in PBS (PBS-T) for 90 min at room temperature. Then, retinas were incubated overnight at 4 ℃ with guinea pig anti-RBPMS (#1832-RBPMS,1:100, PhosphoSolutions, Denver, CO, USA) in blocking buffer. After being fixed in 4% PFA for 10 min and washed three times in PBS-T, the retinas were incubated with the secondary antibody (goat anti-guinea pig Alexa Fluor 488, ab150185, 1:1000, Abcam, Cambridge, UK) in blocking solution for 90 min at room temperature. Finally, retinas were washed in PBS three times and mounted with the mounting medium containing DAPI. Then, retinal whole-mounts were examined with a fluorescence microscope (Leica, Wetzler, Germany). RBPMS-positive RGCs were quantified by two observers using ImageJ software in three areas (center, middle, periphery) in each of the four quadrants of the retina.
Immunofluorescence staining of retinal sections
Retinal cryosections were prepared for immunofluorescence according to standard techniques. Briefly, the mice were killed, and eyes were enucleated and fixed in 4% PFA overnight at 4 ℃. Then, the eyes were incubated in 10%, 20%, and 30% sucrose in PBS and frozen in optimum cutting temperature (OCT) compound (Sakura Finetek, Torrance, CA, USA). Ocular tissue sections were cut through the optic disk of each eye using a cryostat at a thickness of 15 µm and dried on glass slides. Subsequently, cryosections were fixed with 4% PFA for 10 min at room temperature and then blocked with blocking solution (5% BSA in PBS-T) for 30 min. Next, the sections were incubated with the following primary antibodies diluted in the blocking solution overnight at 4 ℃: guinea pig anti-RBPMS (#1832-RBPMS,1:100, PhosphoSolutions, Denver, CO, USA), rabbit anti-RIP1 (#3493, 1:100, Cell Signaling Technology, Danvers, MA, USA), rabbit anti-RIP3 (#95702, 1:100, Cell Signaling Technology, Danvers, MA, USA), and rabbit anti-NLRP3 (#DF7438, 1:100, Affinity Biosciences, USA). Following the primary incubation, sections were stained with the secondary antibodies (goat anti-guinea pig Alexa Fluor 488, ab150185,1:1000, Abcam, Cambridge, UK and goat anti-rabbit Alexa Fluor 647, ab150079, 1:1000, Abcam, Cambridge, UK) for 90 min at room temperature and then mounted with the mounting medium containing DAPI. Images were obtained with a fluorescence microscope (Leica, Wetzler, Germany).
Statistical analysis
All data are presented as mean ± standard deviation (SD) from at least three independent experiments. Statistical analysis was performed using the GraphPad Prism software (version 8.0). Multiple data were analyzed using one-way analysis of variance (ANOVA), followed by Tukey’s multiple comparison test. Student’s t-test was also performed to analyze the differences between the two groups. Differences were considered statistically significant at p < 0.05.





留言 (0)