Overlap syndrome is defined as the coexistence of signs, symptoms, and immunological features of two or more connective tissue diseases that occur simultaneously, sequentially, or at different times in the same patient [1]. Although this concept appears uncomplicated, this rare disease is difficult to diagnose because its genetics and pathogenesis are not completely understood. In the overlap syndrome, mixed connective tissue disease (MCTD) is now treated as a well-defined entity with clearer diagnostic criteria.
Our case fulfilled the diagnostic criteria for MCTD according to the Kasukawa diagnostic criteria. However, we still do not think the patient is a case of MCTD. The main reason is that, nowadays, MCTD is recognized as a distinct entity with unique clinical features, therapeutic response, and prognosis. MCTD is characterized by the concomitant occurrence of clinical symptoms of different rheumatic disorders without meeting the clear diagnostic criteria for these diseases. Our patient met the diagnostic criteria for both SLE and polymyositis. Besides the low positive anti-RNP titer (two-fold ratio related to negative values), the patient had strong anti-dsDNA, anti-Sm, and myositis-specific antibodies, which are rare in MCTD. These results convinced us that the patient had overlap syndrome rather than MCTD [2, 3].
The descriptions of pediatric overlap syndromes are limited to a few case reports and case series [4,5,6]. In pediatric patients initially diagnosed with SLE, the heterogeneity of presentation and course makes the diagnosis of overlap syndrome even more difficult. In clinical practice, the overlap between SLE and systemic sclerosis is more common in East and South Asian populations [1]. Our patient was diagnosed with overlap syndrome with SLE and JPM. The prevalence of overlap between SLE and idiopathic myositis has been reported to be 3.4–6.3% [7, 8]. In a study of adult and pediatric patients with SLE by Bitencourt et al., 6.3% of the patients developed inflammatory myositis. A significantly higher prevalence of overlap myositis was found in childhood SLE patients than in adult patients [9].
At the time of SLE diagnosis, anti-RNP autoantibodies were detected in our patient. Previous reports have shown that the overall disease development in SLE-myositis patients is influenced by the presence of anti-RNP autoantibodies [1, 7, 9]. Therefore, SLE patients with anti-RNP autoantibodies must be monitored meticulously at clinical presentation or follow-up for overlap myositis [1, 9]. Interestingly, as the muscle discomfort and joint symptoms of our patient worsened, her SLE-specific autoantibody levels declined (Table 1). This may warn the clinicians that a patient has developed an overlap.
Our patient developed Raynaud's phenomenon and fingertip vasculitis two years after the initial diagnosis and developed overlap syndrome with SLE and JPM four years after the SLE diagnosis. The overlap syndromes could be diagnosed concurrently with SLE or developed after SLE diagnosis [1, 7, 9], and inflammatory myositis occurred at an average of 5.25 years after SLE diagnosis [9]. In a juvenile case report by Nitta et al., a 16-year-old girl presented with overlap syndrome consisting of SLE from the age of 7 years and JPM from the age of 10, which was later accompanied by systemic sclerosis from the age of 15. However, in this case, Raynaud's phenomenon and pitting ulcers of the fingers and toes occurred one year after the diagnosis of SLE-JPM overlap syndrome, which is different from our case [10].
In contrast to anti-Mi-2 or anti-Jo-1 antibodies, which are more common in autoimmune myositis, our patient tested positive for anti-OJ autoantibodies. Anti-OJ autoantibodies are myositis-specific autoantibodies that target isoleucyl-tRNA synthetase and are associated with anti-synthetase syndrome (ASS) [11]. Anti-OJ autoantibodies can only be found in less than 5% of patients with idiopathic inflammatory myopathies [12]. In addition to arthritis, fever, and Raynaud phenomenon, which occur in ASS, patients with anti-OJ antibodies appear to have more severe myositis. In the study by Noguchi et al., an increased incidence of severe limb and neck muscle weakness, dysphagia, and muscle atrophy was noted in muscle biopsies [11, 13]. Skin involvement may occur in patients who are anti-OJ positive. Patients have been reported to have heliotrope rash, Gottron's sign or papules, V sign, shawl sign, and holster sign. Compared to adult cases, ASS is rare in juvenile disease and seems to cause a lower incidence of ILD in pediatric cases [11,12,13]. Considering the transmission risk during the pandemic COVID-19, a pulmonary function test, especially the single-breath diffusion capacity test, was not performed in our case. The unremarkable HRCT findings in our case imply that there may be differences in the clinical features of positive anti-OJ antibodies between juvenile and adult cases [14].
The gold standard to characterize idiopathic inflammatory myopathies is the morphological, immunohistochemical and immunopathological analysis of muscle biopsy. We did not perform muscle biopsy for three reasons. First, the clinical presentation and the result of MRI revealed that JPM/JDM was highly suspected. Second, autoantibody testing is an important tool for the diagnosis of IIMs. Myositis‑specific autoantibodies such as anti-OJ antibody are almost exclusively present in IIMs. The last reason we did not perform muscle biopsy is that her parents hesitated to do the examination. In fact, a muscle biopsy is not only to confirm IIMs but also helpful to identify the subset of IIMs [15, 16].
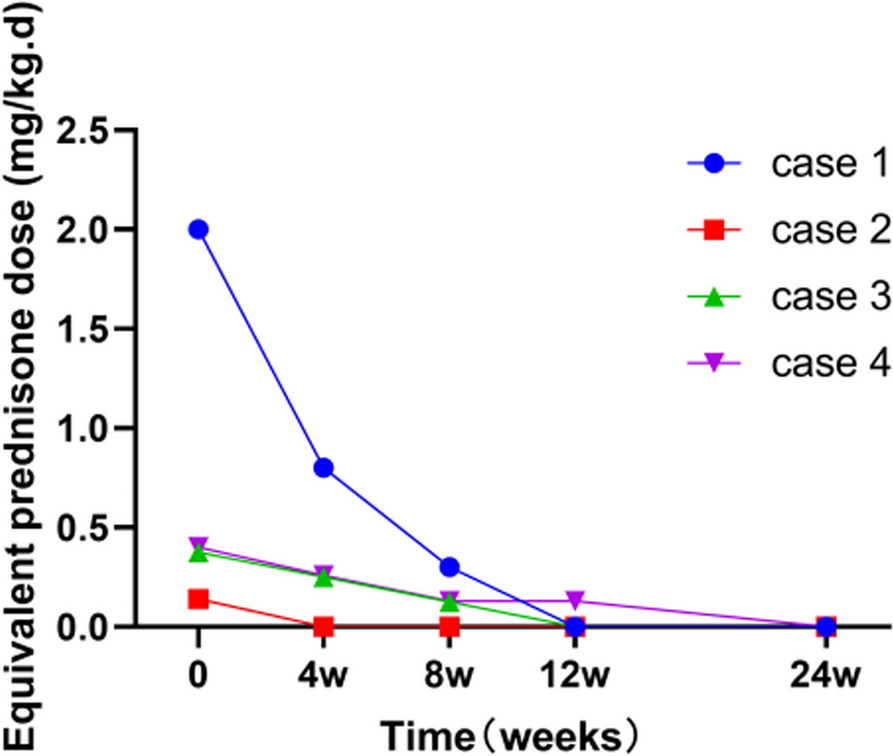
Most patients with anti-OJ have a good response to glucocorticoids [11]. However, patient treatment should be individualized according to different manifestations [17, 18]. Management guidelines are based on clinical manifestations and individual profiles. Therapy for the overlap syndrome usually requires a cocktail of drugs to suppress inflammation. Myositis manifesting clinically as muscle weakness is more common than the laboratory elevation of muscle enzymes. The response to treatment should be mainly evaluated by clinical improvement because, as in our patient, CPK levels do not always correlate with disease activity [19].


留言 (0)