Cells, antibodies, and reagents
HEK293 cells were grown in DMEM medium and ccRCC cell lines (786-O, ACHN, M48, and M62) cells were grown in RPMI/MEM medium supplemented with 100 units/ml of penicillin, 1 μg/ml of streptomycin, and 10% FBS in 5% CO2 incubator at 37 °C. 786-O and ACHN cells were purchased from ATCC and used in 5 passages. To generate SENP1 expressing 786-O cell clones, pCMV-3Flag-SENP1 construct was transfected, and cells were selected by treatment of puromycin (1 μg/mL) for several weeks. Specific clones were isolated by isolating individual colonies. The control cells were also generated in parallel by transfecting 786-O cells with the empty vector, selecting with puromycin and isolating resistant clones as above. For bioluminescence imaging, pGL4-Luc2 (Promega) was transfected into control 786-O cell clone or S1#7 clone and luciferase-expressing stable cell pools were selected with hygromycin B (200μg/mL).
Antibodies used for TMA and immunoblot assays were from Abcam - anti-HIF1α (ab51608), anti-HIF2α (ab109616), anti-SENP1 (ab108981); Sigma Aldrich - anti-Flag (M2): Roche - anti-HA (3F10): and Cell Signaling - anti-Vimentin (D21H3), anti-E-cadherin (4A2), anti-N-cadherin (D4R1H), phospho-AKT (193H12), and phospho-S6K (49D7). Anti-SUMO1 (GMP1) antibody was purchased from Invitrogen and purified from the supernatant of hybridoma cells. For flow cytometry, anti-CD44-FITC (ab19622) was purchased form Abcam. Everolimus was purchased from Selleckchem, LLC.
Public RNA data and tissue microarray (TMA) analyses
To investigate the correlation of expression of HIF’s and SENP1 genes, the publicly available Human Protein Atlas database of RCC samples (N = 877) were analyzed for the correlation (R^2) or covariation by Spearman correlation methods for RNA expression of HIF1α, HIF2α, and SENP1 genes. For tissue microarray (TMA) analysis, malignant and tumor-adjacent benign tissues were used to construct a manual tissue array [43]. A total of 471 malignant cores and 190 benign cores were included. Immunohistochemistry was performed by UW Translational Research Initiatives in Pathology (TRIP) facility. TMA with human samples was performed the protocol (#2011-0179) approved by IRB. Anti-SENP1 (ab108981), anti-HIF1α (ab51608), or anti-HIF2α (ab109616), all from Abcam, MA, was applied to the TMAs, and hematoxylin was used for count-staining. Stained slides were scanned by Vectra slide scanner (PerkinElmer, MA) and SENP1, HIF1α, and HIF2α expression levels in the nuclears (where SENP1 would act on these HIF proteins) were quantified and analyzed in inForm software (PerkinElmer).
In vitro proliferation assay
2 × 104 cells were plated in each well of six-well plates. Cells were grown in the above RPMI culture media, fixed and stained by 2.5% crystal violet solution after 1, 3, or 5 days in culture. The dye was resolved with 50% methanol and measured by a spectrophotometer (540 nm).
In vivo tumor growth and Bioluminescence imaging (BLI)
106 786-O cells (S1#7 or control cell clone) stably expressing luciferase were mixed with 50% Matrigel (BD sciences) and injected into the side flank (subcutaneous injection) of 6–8 weeks old NOD scid gamma (NSG) mice (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ). To measure the growth of injected cells, 2 mg of D-luciferin in 100 μl of 20 mg/ml solution was injected intraperitoneally into each mouse and visualized after 5 min by bioluminescence image (BLI) instrument (IVIS, PerkinElmer). BLI images were taken every 2 weeks for 2 months. There is no randomization and blinding experiments. All animal experiments were performed under a protocol (#M005757) approved by IACUC.
Immunoprecipitation
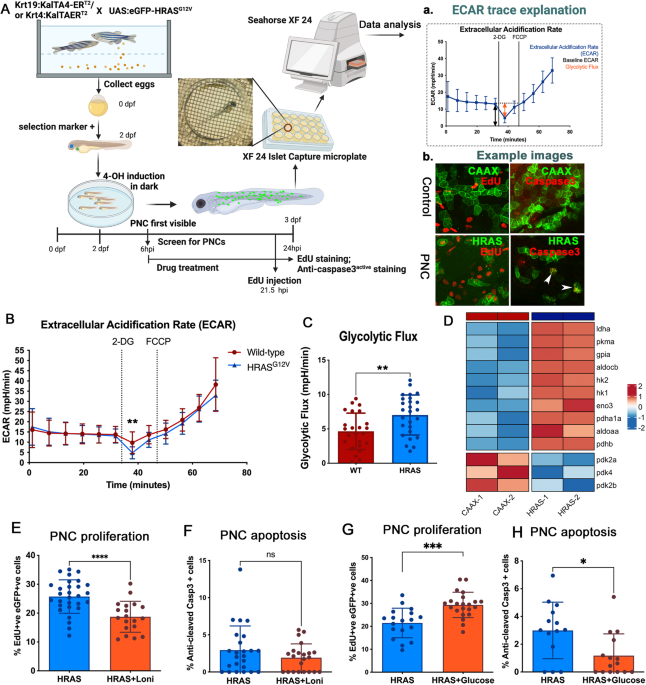
To examine the modifications of HIF2α, cells were lysed by SDS-IP buffer composed of 1% SDS, 3% glycerol and Tris-HCl (pH 6.8), along with 1X protease inhibitor cocktail (Invitrogen). The samples were boiled for 10 min and diluted 10-fold by adding immunoprecipitation (IP) buffer (50 mM Tris-HCl, pH 7.4, 120 mM NaCl, and 0.5 % NP-40). The supernatant was used for IP with anti-HIF2α antibody and sumoylation of HIF2α was analyzed by immunoblotting with anti-SUMO1 antibody (GMP1). For co-immunoprecipitation of SENP1 and HIF1α or HIF2α, HEK293 cells were transfected with HA-tagged HIF1α, HA-tagged HIF2α, or Flag-tagged SENP1 vectors, and cell lysates were made at 24 h after transfection. Anti-Flag antibody (M2) was used to immunoprecipitate Flag-SENP1 complexes and anti-HA and flag antibodies were used for immunoblot analysis. Band intensities were quantified by ImageJ software.
RNA-seq and qRT-PCR analyses
Total RNAs were prepared by Nucleospin RNA kit (Macherey-Nagel Inc, PA). After reverse transcription, the resulting cDNA was used for quantitative RT-PCR experiments using the primers described in Supplementary Fig. 7. RNA-seq integrated workflow service was provided by ProteinCT Biotechnologies (Madison, WI). For library preparation, total RNA was isolated from 786-O vector control (clone #1 and #2) and SENP1-overexpressing cells (clone #7 and #9) using TRIZol reagent (Life Technologies), and mRNA libraries were prepared using the Illumina TruSeq strand-specific mRNA sample preparation system (Illumina). The libraries were sequenced (Single end 100 bp reads) using the Illumina HiSeq4000, a final of around 30–40 million reads per sample. The fastQC program was used to verify the raw data quality of the Illumina reads. The hg19 human genome and Ensembl gene annotations (v75) were used for mapping. The raw sequence reads were mapped to the genome using Subjunc aligner from Subread, with the majority of the reads (over 96% for all samples) aligned to the genome. The alignment bam files were compared against the gene annotation GFF file, and raw counts for each gene were generated using the feature. Counts tool from Subread, with 84–87% of reads overall assigned to genes. The raw counts data were normalized using voom method from the R Limma package, then used for differential expression analysis. The normalized data were analyzed for their clustering groups by GSEA and MSigDB software (UCSD and Broad Institute) [44].
Gelatin zymography
105 cells were plated in each well of 6-well plates and the growth media were replaced with the same media without serum the next day. After 24 h, the conditioned media were collected and subjected to 0.1% gelatin/SDS-PAGE under non-reducing conditions and without sample boiling prior to gel elecrtophoresis. The gel was renatured by incubating in Tris-HCl buffer (50 mM Tris-HCl pH7.4) and incubated in reaction buffer (50 mM Tris-HCl pH7.4, 200 mM NaCl, and 10 mM CaCl2) for 16 h, at 37 °C. After incubation, the gel was stained with Coomassie R-250 solution and incubated with destaining buffer (10% Acetate, 40% Methanol, and 50% water). The clear bands were used for the measurement of activity of MMP2 or MMP9 by ImageQuant program.
Quantification of secreted MMPs
To measure the amount of secreted MMPs, 106 786-O cells were incubated with serum-free RPMI1640 media for 24 h. The collected conditioned media were incubated with the mixture of microbeads conjugated specific antibodies, such as MMP1/2/7/9/10. After incubation for 1 h, the samples followed washing steps as per the manufacturer’s protocols. The beads previously dyed with distinct spectral sets were measured individually by an xMAP instrument (Luminex, IL, USA).
In vitro invasion assay
2000 cells were mixed with collagen (1 mg/ml) and 25% Matrigel in serum-free RPMI1640 media. Cells were loaded to microchannels for invasion assay as described in refs. [45, 46]. This microchannel is designed for invasion assay and has 2 channels linked to a microchannel between them. One side was loaded with cells in gel and the other side was loaded with complete growth media for inducing cell movement by chemotaxis. After 3 days, cells were fixed with 4% PFA and stained with phalloidin-rhodamine (Abcam, MA), and migrated cells out of the gels were counted manually using a microscope.
In vivo tumor invasion assay
For in vivo invasion assay, 3.5 × 105 above cells were mixed with neutralized collagen and placed under kidney capsule (orthotopic injection) of 6–8 weeks old NSG mice. After 2 months, kidneys were isolated and analyzed by H&E staining for invasion of ccRCC tumors.
Sphere forming assay
For the growth of cancer stem cells, 200 cells (luc-expressing 786-O) were plated in each well of 24-well low-attachment plates (Nunc) with sphere-forming media (MEM media supplemented with 1% penicillin-streptomycin, 20 ng/ml human EGF, 20 ng/ml human basic FGF, and 1X B27) [47]. Cells were supplied with fresh media every three days for 10–14 days. Visible spheres were counted under a microscope and the lysate from these spheres was measured for the luciferase activity by a luminometer.
Drug toxicity assay
2 × 104 luciferase-expressing 786-O cells were seeded in 96-well plate with various doses of drugs (0–1.5 μM). After 24 h, cells were harvested, and luciferase activity was measured using the substrate luciferin.



留言 (0)