
記住我
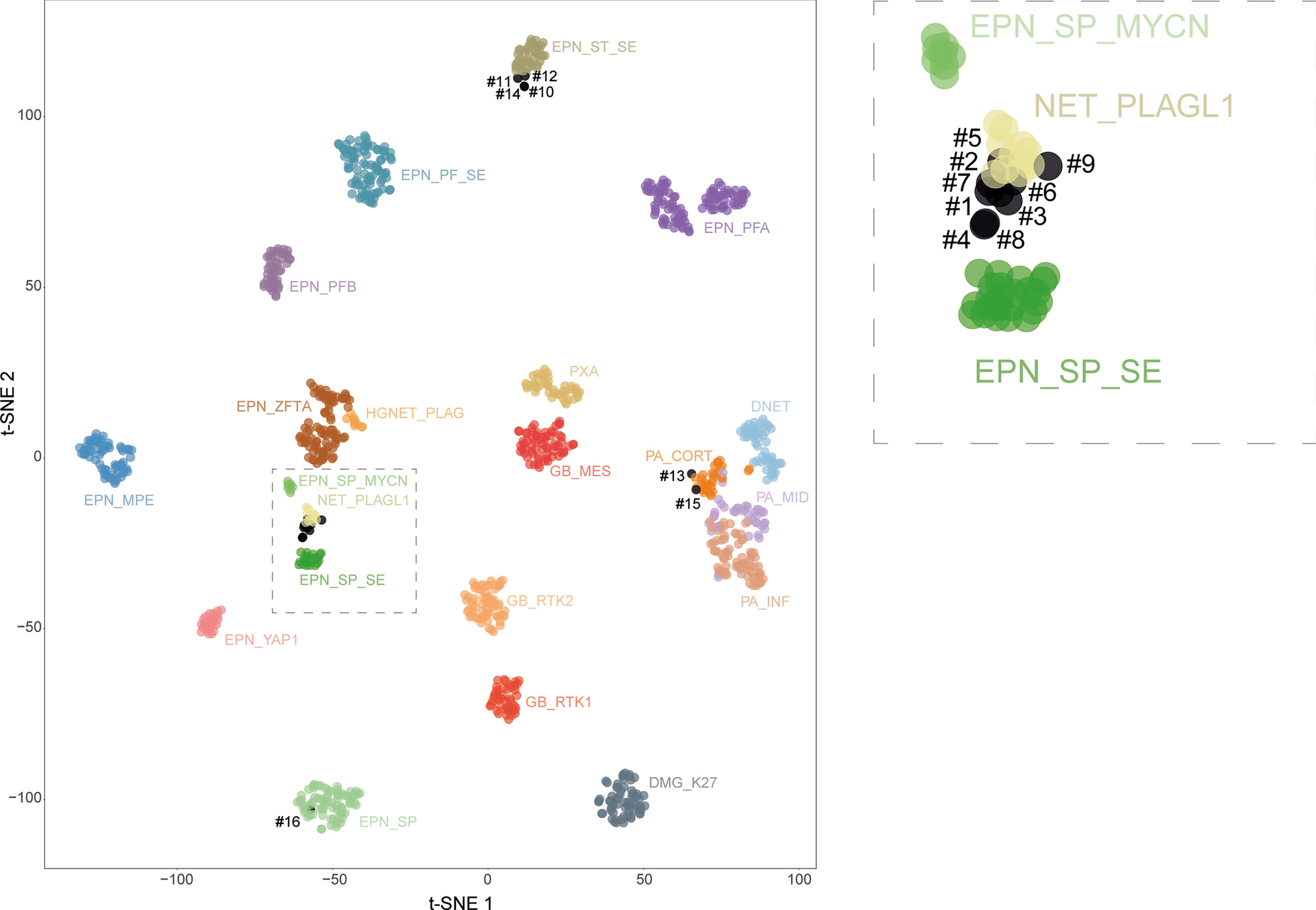
We measured the plasma concentration of soluble CD146 (sCD146) in a cohort of 17 patients with recurrent IDHwt glioblastoma who were treated for 3 weeks with bevacizumab and carmustine (Fig. 1a). Plasma samples were collected just before the second bevacizumab administration. Responses after bevacizumab-chemotherapy administration were either complete (1 patient) or partial (5 patients), corresponding to responders, or stable (4 patients) or progressive (7 patients), corresponding to non-responders. We show that sCD146 plasma level, under treatment, was higher in non-responder patients than in responders (Fig. 1b), and that this corresponded to an increase in sCD146 occurring after treatment (Fig. 1c). Importantly, a high sCD146 plasma level in patients treated with one cycle of bevacizumab significantly associated with shorter PFS (p = 0.049) and OS (p = 0.032) (Fig. 1d). Patients with a high sCD146 plasma concentration had a median PFS of 3.0 months (95% CI 2.5–3.6) as compared to 5.2 months (95% CI 3.4–6.9) in patients with low sCD146 plasma concentration. Patients with a high sCD146 plasma concentration had a median OS of 6.1 months (95% CI 3.7–8.6) versus 9.6 months (95% CI 4.8–14.3) in patients with low sCD146 plasma concentration. In contrast, neither age, Karnofsky score (KPS) nor steroid dose were associated with patient PFS or OS in univariate analyses (Additional file 1: Table S1), suggesting the independence of sCD146 from classical prognostic factors.
Fig. 1
sCD146 plasma concentration in patients under bevacizumab treatment. a Patients’ characterization. b sCD146 plasma concentration in patients after one cycle of bevacizumab treatment (Bev) between responders and non-responders. c Evolution of sCD146 plasma concentration at baseline versus after one cycle of bevacizumab treatment, in responder and non-responder patients. d Progression-Free survival (left) and Overall survival (right) according to sCD146 plasma concentration in patients under bevacizumab treatment. *p < 0.05, non-responders versus responders
These results highlight the pejorative impact of sCD146 increase in patients treated with bevacizumab and its potential implication in bevacizumab resistance.
Long-term treatment with Bevacizumab significantly induces CD146/soluble CD146 signaling pathway in CD146-positive glioblastoma cell linesIn this study, we used 3 different glioblastoma cells lines, U87, U373 and U118. Flow cytometry analysis of U87 and U373 glioblastoma cell lines demonstrated potent expression of CD146, VEGFR2 and integrin αvβ3 in these two cell lines. In contrast, U118 cells did not express integrin αvβ3 and VEGFR2, only faintly expressed CD146, and were thus used as a negative control (Additional file 1: Fig. S2). The secretion capacity of sCD146 and VEGF from the three glioblastoma cell lines was determined in vitro. ELISA experiments on culture supernatant evidenced a high secretion of sCD146 in U87 and U373 cell lines while U118 cells did not secrete sCD146. In contrast, U87, U373 and U118 highly secreted the Vascular Endothelial Growth Factor (VEGF) (Additional file 1: Fig. S3).
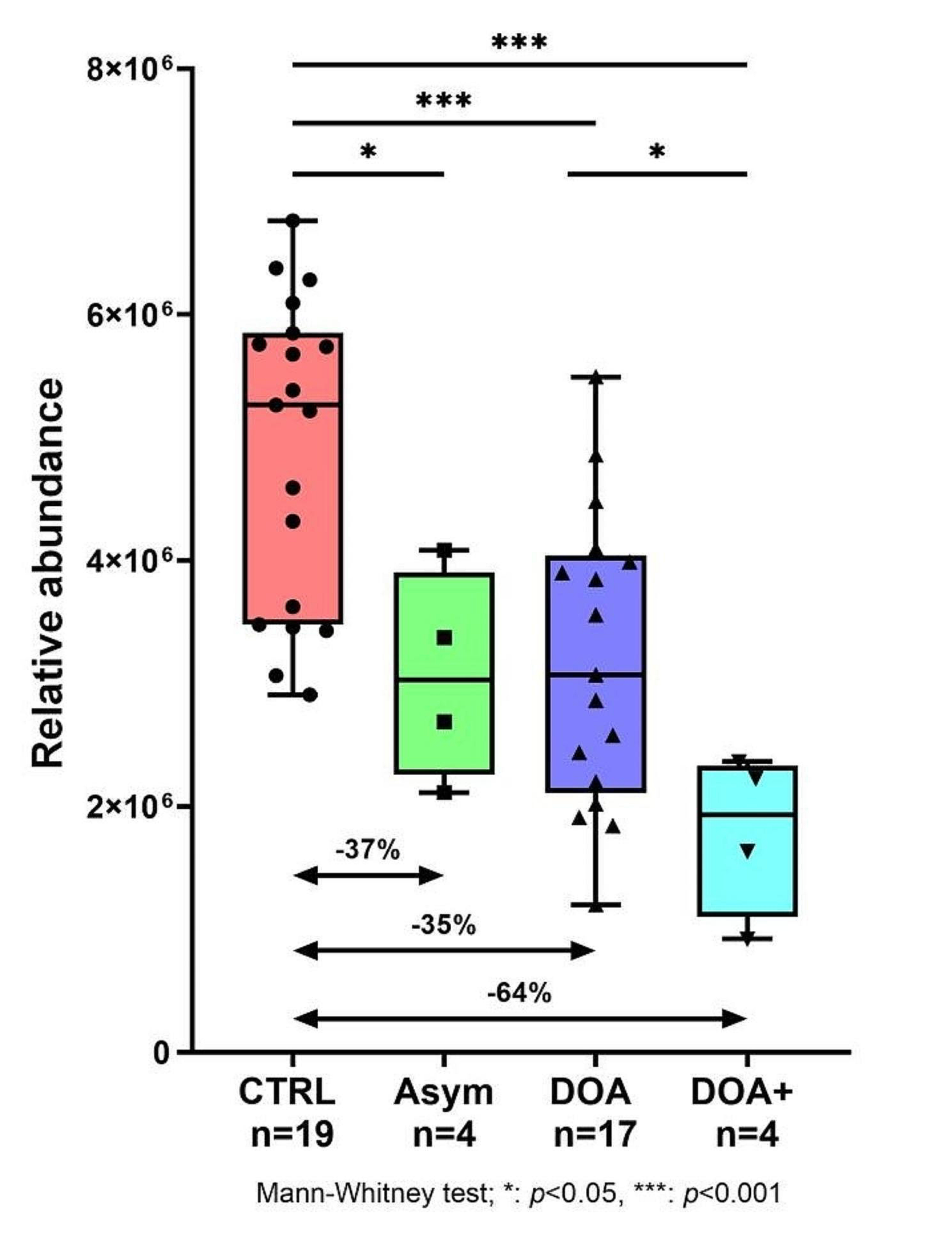
To generate Bevacizumab-resistant GBM cell lines, we treated U87, U373 and U118 for two months with 100 µg/ml bevacizumab (commercially known as Avastin) or irrelevant IgG as a control. The absence of antibody toxicity was confirmed using trypan blue exclusion assay and live/dead fixable aqua staining dye (data not shown). After 2 weeks of treatment, we noticed a robust increase in U87 and U373 cell proliferation, but not in U118 cell proliferation, an effect that was maintained during the entire treatment course (Fig. 2a; Additional file 1: Figs. S4A; S5A). In addition, a significant increase in CD146, VEGFR2, integrin subunits αv and β3 at the mRNA levels was detected in U87 and U373 cell lines, but not in U118 cells (Fig. 2c; Additional file 1: Figs. S4C; S5C). As membrane CD146 expressed at the membrane can be shed to generate sCD146, we analyzed the membrane expression of these proteins by flow cytometry. The membarne expression of integrin αvβ3 and VEGFR2 were increased but CD146 membrane expression decreased (Fig. 2d; Additional file 1: Fig. S4D). In contrast, membrane expression of the three surface proteins did not change in U118 cells (Additional file 1: Fig. S5D).
Fig. 2
Bevacizumab induces proliferation and CSC/EMT markers in U87 cells. In-vitro challenging of U87 cells with bevacizumab (Avastin; 100 µg/ml) for 2 months enhanced cell proliferation (a) and sCD146 secretion (b), normalized to total cell number, as compared to IgG treated cells. Avastin-challenged U87 cells upregulated CD146, VEGFR2 and integrin subunits αv and β3 gene transcription (c) and their membrane expression, except for CD146 (d). U87 challenging with Avastin potently induced expression of markers related to CSC (e) and EMT (f) at the mRNA level as compared to control IgG treated cells. Average of 5 experiments is shown; **p < 0.01, ***p < 0.001, experimental vs control
Besides, sCD146 concentration was significantly increased in the culture media of U87 and U373 cells after exposure to bevacizumab as compared to irrelevant IgG (Fig. 2b; Additional file 1: Fig. S4B). This increase in sCD146 concentration was not detected in the supernatant of bevacizumab-treated U118 cells (Additional file 1: Fig. S5B).
We then examined Epithelial-Mesenchymal Transition (EMT) and Cancer Stem Cells (CSC) markers in the bevacizumab-challenged glioblastoma cells. We analyzed the main CSC markers oct4, sox2 and nanog, and the EMT markers that are commonly expressed in GBM cells, snail, slug, vimentin, N-cadherin, S100A4, Twist1, and Zeb1, at the mRNA level. All these markers were potently upregulated in the bevacizumab challenged U87 cell line as compared to IgG treated cells (Fig. 2e and f). Similar results were obtained in the U373 cell line (Additional file 1: Fig. S4 E–F) but not in the CD146-negative glioblastoma cell line U118 (Additional file 1: Fig. S5 E–F).
Soluble CD146 reproduces the effects observed with long-term bevacizumab treatment in CD146-positive glioblastoma cellsAs bevacizumab treatment of CD146-positive (CD146 +) glioblastoma cells led to a potent increase in sCD146 secretion, we investigated sCD146 effect on U87 and U373 cells. Our results show that in vitro stimulation of these cells for 48 h with recombinant sCD146 or VEGF significantly increased cell proliferation, migration, and invasion. An additive outcome effect was achieved when combining both molecules, in comparison to VEGF or sCD146 treatment alone (Fig. 3a–c; Additional file 1: Fig. S6 A–C). In contrast, in U118 cells, sCD146 or VEGF failed to induce cell proliferation (Additional file 1: Fig. S7).
Fig. 3
Soluble CD146 induces U87 cell proliferation, migration and invasion and promotes CSC/EMT markers. U87 cells were treated with 100 ng/ml of VEGF, rsCD146 or combination of both molecules for 48 h and cell proliferation (a), migration (b) and invasion (c) were determined. EMT (d) and CSC (e) markers were also examined after 48 h of treatment with 100 ng/ml sCD146. Representative blots from 5 experiments are shown. *p < 0.05, **p < 0.01, ***p < 0.001, experimental vs control
Moreover, sCD146 potently induced the expression of the mesenchymal markers snail, slug, N-cadherin and vimentin, but decreased the epithelial marker E-cadherin at the protein level in U87 cells. Likewise, sCD146 upregulated the expression of the cancer stem cell markers nanog, oct4 and sox2 in U87 cells (Fig. 3d and e). Similar results were obtained in U373 cell lines (Additional file 1: Fig. S6 D and E).
Soluble CD146 mediates its effects through integrin αvβ3 in CD146-positive glioblastoma cellsIn a previous work, we showed that sCD146 interacts with angiomotin on endothelial cells to promote angiogenesis [10]. Thus, we examined angiomotin expression on U87, U373, and U118 cells. Flow cytometry analysis showed a weak expression of this surface protein (Fig. 4a; Additional file 1: Fig. S2). In contrast, integrin subunits αv and β3 were expressed by U87 and U373 but not U118 cells as assessed by RT-PCR and flow cytometry. Therefore, we tested a possible interaction between sCD146 and integrin αvβ3. Silencing RNA targeting integrin subunits αv or β3 significantly reduced sCD146-induced increase in proliferation of U87 and U373 cells, whereas siRNA targeting other integrin subunits, αL or β2, did not modify cell proliferation (Fig. 4b; Additional file 1: Fig. S8A). Moreover, knocking down integrin αvβ3 using silencing RNA targeting either αv or β3 subunits, but not αL or β2 integrins, on U87 and U373 cells, led to a significant decrease in sCD146-FITC binding to these cells (Fig. 4c; Additional file 1: Fig. S8B). In order to confirm the specific binding of sCD146 on αvβ3, we transfected Chinese Hamster Ovary cells (CHO), which originally do not express this integrin, with αvβ3. Exogenous expression of integrin subunits αv and β3 into CHO cells induced a significant increase in sCD146-FITC binding to these cells as revealed by flow cytometry and immunofluorescence experiments (Fig. 4d and e). In addition, anti-αvβ3 antibody immunoprecipitated sCD146 only when CHO cells expressed this integrin after sCD146 treatment. To confirm the specificity of the immunoprecipitated sCD146, we treated CHO cells with blocking anti-αvβ3 antibody before adding sCD146. Results show that anti-αvβ3 antibody potently reduced the interaction with sCD146 (Fig. 4f).
Fig. 4
Soluble CD146 binds integrin αvβ3 on U87 cells. The expression of angiomotin (Amot) and αvβ3 on U87 cells was analyzed by flow cytometry (a). The effect of silencing RNA targeting αv, β3, αL and β2 was analyzed on sCD146-induced U87 cell proliferation (b). U87 cells were transfected with siRNA targeting αv, β3, αL and β2 and sCD146-FITC binding was determined by flow cytometry (c). CHO cells were co-transfected with plasmids encoding integrin subunits αv and β3 or control plasmids (mock) and sCD146-FITC binding was determined by flow cytometry (d) or by immunofluorescence microscopy (e). Mock transfected or αvβ3 expressing CHO cells were treated or not with blocking anti-αvβ3 antibody and then sCD146 was added for 1 h at 37°. CD146 was examined after immunoprecipitation with anti- αvβ3 antibody. Loading was analyzed using IgG heavy chain (f). In two ELISA assays, the effect of cyclic RGD peptide/mucizumab (g) and the dissociation constant Kd (h) were estimated for the binding of αvβ3 to rsCD146. White bars correspond to 25 µm. Average of 3 experiments is shown; *p < 0.05, **p < 0.01, ***p < 0.001, experimental vs control
In-house ELISA also confirmed a strong interaction between integrin αvβ3 and sCD146 with an estimated dissociation constant of 1.8 nM. The addition of blocking cyclic RGD peptide failed to inhibit this interaction even at high concentrations, in contrast to a newly generated humanized neutralizing monoclonal anti-sCD146 antibody, mucizumab, which was generated by our laboratory (Fig. 4g and h).
Finally, we overexpressed or silenced integrin αvβ3 in U118 and U87 cells and studied EMT markers in response to sCD146 stimulation. Results showed that only cells expressing αvβ3 upregulate EMT markers when treated with sCD146 (Additional file 1: Figs. S9; S10).
CD146 is part of a signalosome containing VEGFR2 and Integrin αvβ3To analyze the components of the signalosome containing CD146, we performed co-immunoprecipitation experiments using anti-CD146 antibody, S-Endo1. Immunoblotting with anti-VEGFR2, anti-αv, and anti-β3 antibodies revealed specific bands corresponding to these proteins. Membrane probing with an antibody to another surface protein, EPCAM, failed to detect it, attesting the specificity of the immunoprecipitated proteins (Additional file 1: Fig. S11 A). Results were reproduced in U373 cell line (Additional file 1: Fig. S12 A).
As CD146 is a co-receptor for VEGFR2, and since VEGFR2 activation induces its own phosphorylation at multiple tyrosine residues [11], we investigated whether sCD146 or VEGF induces the phosphorylation of CD146 and whether integrin αvβ3 is indispensable in this process. Accordingly, U87 cells were treated with either sCD146 or VEGF, and CD146 was immunoprecipitated followed by western blotting using anti-pan phospho-tyrosine antibody. Results showed that sCD146 and VEGF increased the phosphorylation of membrane CD146 (Additional file 1: Fig. S11 B). Next, we knocked-down integrin αvβ3 using silencing RNA and reproduced the experiment. A potent decrease in CD146 phosphorylation in response to sCD146 or VEGF stimulation was detected as compared to control cells transfected with scrambled RNA (Additional file 1: Fig. S11 B). These results were reproduced in U373 cells (Additional file 1: Fig. S12 B).
We then studied the phosphorylation of integrin β3. Our results show a significant increase in integrin β3 phosphorylation in response to sCD146 or VEGF stimulation (Additional file 1: Fig. S11 C). To examine if CD146, integrin αvβ3, or VEGFR2 are crucial in the phosphorylation and activation of the newly defined signalosome, we used CRISPR-Cas9 technology to individually knockout each of these proteins (Additional file 1: Fig. S13). We showed that the knockout of CD146 on U87 cells significantly reduced integrin β3 and VEGFR2 phosphorylation following sCD146 or VEGF stimulation. Also, knocking out integrin β3 on U87 cells significantly reduced or abolished VEGFR2 phosphorylation in response to VEGF or sCD146 stimulation (Additional file 1: Fig. S11 D). In contrast, VEGFR2 knockout on U87 cells abrogated integrin β3 phosphorylation in response to VEGF stimulation but not sCD146 (Additional file 1: Fig. S11 C). Similar results were obtained in U373 cells (Additional file 1: Fig. S12 C-D).
Of interest, we also showed this signalosome to exist in human umbilical endothelial cells, HUVEC, and revealed that sCD146, through integrin αvβ3, induces cell proliferation and migration (Additional file 1: Fig. S14).
These results demonstrate a complex constituted by the surface proteins CD146, VEGFR2 and integrin αvβ3 that mediates sCD146 and VEGF effects on CD146 + glioblastoma cells, but also on endothelial cells.
Soluble CD146 and VEGF activate common signaling pathways in CD146-positive glioblastoma cell linesWe next investigated the signaling pathways that are implicated in mediating sCD146 and VEGF effects in CD146 + glioblastoma cells. To this end, we studied the phosphorylation of key proteins involved in major signaling pathways regulating cell survival, proliferation, and invasion. Our results show that, similar to VEGF, sCD146 induced the phosphorylation of P38 MAPK, AKT, and ERK44/42 proteins. Besides, both sCD146 and VEGF induced the phosphorylation and activation of focal adhesion kinase (FAK). The phosphorylation of these proteins became significantly impaired when either CD146, VEGFR2, or integrin αvβ3 were knocked out (Additional file 1: Figs. S15 and S16). These effects were functionally translated to a decrease in the ability of cells to proliferate and migrate in response to sCD146 and/ or VEGF stimulation (Additional file 1: Figs. S17 and S18).
Thus, CD146, VEGFR2 and integrin αvβ3 interact together to translate extracellular stimuli into intracellular signals, and each of these proteins is required to potentiate the activated signaling pathways.
A novel humanized anti-sCD146 antibody, mucizumab, blocks sCD146 adverse effects on CD146-positive glioblastoma cells and prevents escape from bevacizumab in vitroIn order to block glioblastoma escape from bevacizumab, we tested the effect of the humanized neutralizing monoclonal anti-sCD146 antibody, mucizumab. The addition of 100 µg/ml of either bevacizumab, mucizumab, or the combination of both antibodies to U87 or U373 conditioned media (CM) resulted in a significant decrease in cell proliferation, migration, and invasion in vitro. Our results showed that mucizumab displayed greater effects than that achieved by bevacizumab, while the combination of mucizumab and bevacizumab significantly extended the inhibitory effects of bevacizumab on cells’ proliferation, migration, and invasion as compared to bevacizumab treatment alone (Fig. 5a–c; Additional file 1: Fig. S19 A-C).
Fig. 5
Humanized anti-sCD146 antibody mucizumab significantly decreases U87 cell proliferation, migration and invasion and hampers CSC and EMT in vitro. U87 cells were treated with conditioned media (CM) containing Irrelevant IgG, bevacizumab (Avastin), Mucizumab, or combination of both antibodies for proliferation (a), migration (b), and invasion assays (c). EMT and CSC markers were also examined at the mRNA (d and e) and protein (f and g) levels. Average of 3 experiments is shown; *p < 0.05, **p < 0.01, ***p < 0.001, experimental vs control
We then reproduced the long-term bevacizumab challenging experiment and added mucizumab to the media along with bevacizumab. The effect on EMT and CSC induction in U87 and U373 cells was evaluated. Our results showed a robust decrease in the tested EMT and CSC markers when mucizumab was added to bevacizumab (Fig. 5d–g; Additional file 1: Fig. S19 D-E).
These data confirm that CD146 + glioblastoma cells escape bevacizumab through sCD146 secretion.
Combination of bevacizumab with mucizumab exhibits greater inhibitory effects than bevacizumab alone on tumor growth in different preclinical modelsTo investigate the in vivo relevance of combining bevacizumab with mucizumab, U87 cells were xenografted subcutaneously in athymic nude mice and tumor growth was weekly monitored with caliper. After 5 weeks of treatment, results showed that mice receiving mucizumab developed significant smaller tumors than those receiving IgG. Of importance, an additive inhibitory effect on tumor growth was detected when bevacizumab was combined with mucizumab (Fig. 6a). We measured the concentrations of human VEGF (hVEGF) and human sCD146 (hsCD146) in the plasma of treated mice. Our results showed that the combination of bevacizumab and mucizumab significantly decreased hVEGF and hsCD146 concentrations in the plasma from treated mice as compared to IgG-control group whereas bevacizumab alone increased hsCD146 (Additional file 1: Fig. S20 A-B).
Fig. 6
Humanized anti-sCD146 antibody, mucizumab, significantly reduces tumor growth in two different in-vivo models and displays complementary effects with bevacizumab. Tumor volume was measured in 20 athymic nude mice subcutaneously injected with U87 cells after treatment with IgG, bevacizumab (Avastin), mucizumab or bevacizumab + mucizumab. 5 mice were used in each group. Representative images of tumors and their weight are shown (a). 40 mice were orthotopically injected with U87 cells and treated with IgG, Avastin, mucizumab or bevacizumab + mucizumab. 10 mice were used in each group. Tumor volume was estimated based on immunohistochemical staining of human CD146 in serially cut mice brain sections (b). Tumor cell dissemination across all the brain was also determined. Red arrows show disseminated cells (c). Expression of human specific CD146 and MAX proteins in total brain lysate was determined in each group (d). In each group, imaging was performed in 6 representative mice orthotopically bearing U87 tumors using PET-scan after 86 Ga-RGD injection. Results were expressed as ratio of radioactivity between right (RH) and left (LH) hemispheres (e). Representative images are shown. *p < 0.05, **p < 0.01, ***p < 0.001, experimental vs control
We also performed another in vivo study by orthotopically injecting U87 cells in nude mice. To evaluate the therapeutic benefit of the administered antibodies, immunohistochemistry was done to estimate tumor growth. Similar to that in ectopic tumor model, mucizumab significantly reduced tumor volume and metastasis, while the combination of mucizumab and bevacizumab greatly enhanced these effects. In contrast, bevacizumab, as a monotherapy, did not decrease intracranial tumor growth but rather increased tumor dissemination across the brain parenchyma (Fig. 6b and c). In addition, immunoblots using brain lysate from treated mice showed that the human proteins CD146 and MAX (a DNA-binding transcriptional regulator used as a human protein internal control) decreased when treating with bevacizumab and mucizumab to reach barely detectable levels when both antibodies were combined as compared to IgG-treated group (Fig. 6d). This indicates a large decrease in human tissue corresponding to GBM after treatment with both antibodies. This was confirmed by PET imaging showing a slight significant decrease of the tumor when bevacizumab was used, whereas a large decrease was observed in the presence of both bevacizumab and mucizumab. Along this line, the decrease was significantly higher in the bevacizumab + mucizumab group, as compared to bevacizumab group (Fig. 6e). Also, we showed a significant decrease in plasma concentration of hVEGF and hsCD146 in mice receiving simultaneously bevacizumab and mucizumab antibodies as compared to IgG-control group (Additional file 1: Fig. S20 C-D).
These results emphasize the interest of combining anti-VEGF and anti-sCD146 therapy for achieving maximal inhibitory effects on glioblastoma growth.
留言 (0)