
記住我


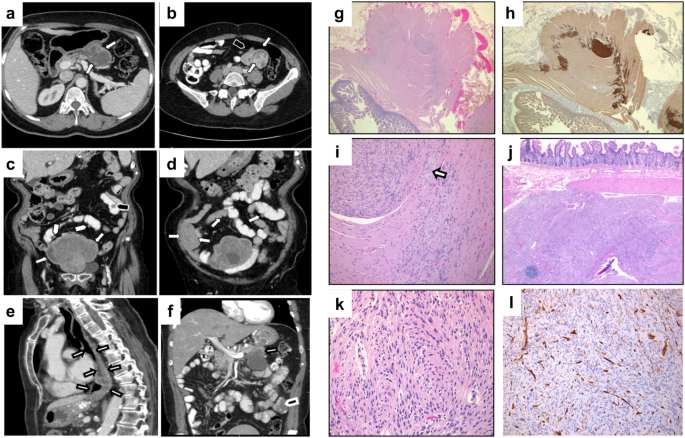
Study individuals underwent virtual gene panel analysis for candidate variants in genes previously shown to be associated with posterior segment disorders, no variants were identified. Subsequent genome-wide analysis identified candidate biallelic variants in CoQ10 biosynthesis pathway genes in 13 affected individuals with a clinical diagnosis of retinitis pigmentosa (RP) from 12 unrelated families (Fig. 1). Biallelic variants were identified in PDSS1 (6 families, 6 affected individuals), COQ2 (3 families, 4 affected individuals), COQ4 (1 family, 1 affected individual) and COQ5 (2 families, 2 affected individuals) genes (Fig. 1, Table 1) in patients with simplex or syndromic RP (Table 2). All affected individuals had vision problems in the dark or low light conditions, constricted visual fields and relatively preserved central vision. Some of the affected individuals were diagnosed with RP later in life following referral at a routine vision check-up. Fundus examination showed retinal vessel attenuation and bone spicule pigmentation in the mid-periphery (Fig. 2). Fundus autofluorescence showed corresponding retinal pigment epithelium (RPE) atrophy in the mid-periphery with a ring of hyperautofluorescence at the macula delineating the border between functional (inside the ring) and non-functional retina (outside the ring). OCT imaging showed loss of photoreceptor outer segments in the outer macula, corresponding to the hyperautofluorescent ring, loss of RPE, as well as decreased choroidal thickness, cystoid macular oedema and epiretinal membrane in some of the affected individuals (Fig. 2 and Supplementary Fig. 1). Various degrees of hearing impairment (4/12, 33.33%) and cardiac problems (4/12, 33.33%) were the most common extraocular features observed in the study cohort. Two affected individuals carrying biallelic pathogenic variants in COQ2 were diagnosed with renal failure, which required renal transplantation. One of them, II:1 from family-7 presented with childhood onset renal failure, which required renal transplantation at the age of 7 years and re-transplantation at the age of 30 years. For affected individual II:5 from family-9 renal transplantation was performed at the age of 55 years. A summary of clinical manifestations identified in the study cohort is presented in Table 2.
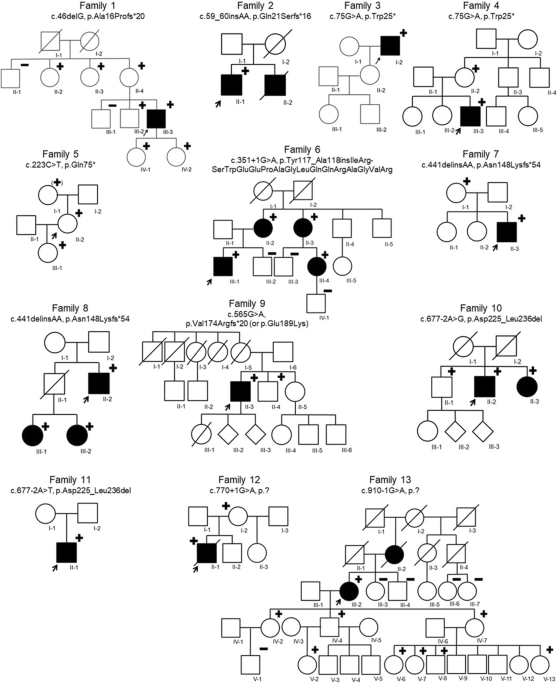
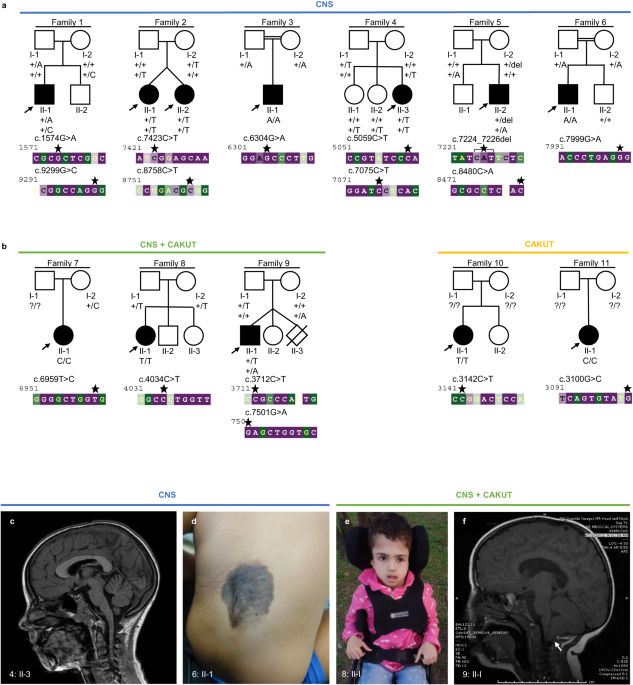
Fig. 1: Pedigrees of families and identified candidate pathogenic variants in CoQ10 biosynthesis pathway genes.
An arrow indicates the proband. Shaded symbols represent affected individuals. Different shading in family 9 indicates unrelated vision loss (not retinopathy). M – mutant, WT – wildtype.
Table 1 Genotypes identified in study cohort.Table 2 Clinical manifestations of study patients.Fig. 2: Multimodal imaging composite of control (right eye only) and four affected individuals (left eye only) carrying candidate pathogenic variants in PDSS1, COQ2, COQ4 and COQ5.
Ultra-widefield fundus pseudocolor imaging shows characteristic features of retinitis pigmentosa: retinal vessels attenuation, pigmentary (bone spicule) changes in mid-periphery. Ultra-widefield fundus auto-fluorescence image demonstrates hypoautofluorescence corresponding to mid-peripheral bone spicules with a hyperautofluorescence ring at the macula delineating the border between normal (inside the ring) and abnormal retina (outside the ring). SD-OCT macula scan through the fovea shows absence of inner segment/outer segment (IS/OS) junction (red arrow) in the parafoveal region in affected individuals carrying candidate pathogenic variants in PDSS1, COQ2 and COQ5. Absence of IS/OS junction in the fovea and parafoveal region in affected individual from family 10, II:5.
Molecular genetic analysisNine distinct variants in PDSS1 (NM_014317.5, Table 1, Fig. 1), of which two, c.18 G > A, p.(Trp6*) and c.893dup, p.(Asn298Lysfs*11), were considered to be probable loss-of-function (LOF). Two variants were predicted to cause splice site disruption: c.722-2 A > G and c.468-25 A > G (Table 3). The variant c.589 A > G, p.(Lys197Glu) was identified in three affected individuals from three unrelated families (Table 1, Fig. 1) and in trans with a probable LOF or splice disrupting variant. This variant is present in 411/282764 alleles (MAF 0.001454) in gnomAD and a maximum allele frequency of 0.003499 is observed within the Latino/Admixed American population, with a single homozygote observed (gnomAD v3). The variant has 6 submissions to the ClinVar database (Benign (1 record), Likely benign (3 records), Variant of uncertain significance (2 records)). The Odds Ratio (OR) that PDSS1 c.589 A > G, p.(Lys197Glu) is associated with disease is 108.34 (Fisher’s exact p = 4.699 × 10−6, 95% confidence interval: 20.47, 374.14. Supplementary Table 1). In light of the recurrence of this variant in trans with probable LOF variants, we considered this to be a candidate pathogenic genotype. No affected individual carried biallelic PDSS1 null variants. Two patients carried biallelic missense or inframe indel variants that are considered to be possibly pathogenic at present (families 2 and 4, variants c.232 C > A, c.886 G > A, c.1118_1120dup).
Table 3 In silico prediction on aberrant splicing.Three different variants were identified in COQ2 (NM_015697.9) with a previously reported variant c.683 A > G, p.(Asn228Ser) observed in all affected individuals from three unrelated families (Table 1). Haplotype analysis (data not shown) showed no shared single nucleotide variants (SNV) in cis with the variant in different families suggesting the variant to have arisen independently. Only one COQ2 probable LOF variant, c.735 C > G, p.(Tyr245*) was identified, and this was in the individual observed in this study cohort to have childhood onset kidney disease.
One study subject was identified to carry two previously unreported missense variants in COQ4 (NM_016035.5): c.692 G > A, p.(Cys231Tyr) and c.376 G > A, p.(Glu126Lys). No other candidate genotype was identified in any known disease gene following GS for this individual, therefore we consider the COQ4 genotype to be possibly pathogenic in this case.
Three distinct variants were identified in the COQ5 gene (NM_032314.4). A candidate non-canonical splice site variant (c.682-7 T > G) was identified in two unrelated individuals from families 11 and 12 in trans with a frameshift variant, c.933delC p.(Tyr311*), and missense, c.367 C > T p.(Arg123Trp), respectively.
All variants except PDSS1 c.589 A > G, p.(Lys197Glu) and COQ5 c.682-7 T > G were rare or absent from the gnomAD dataset (MAF <0.001, Table 1). The in silico algorithm, Mutation Taster, predicted all protein coding variants to be ‘disease causing’ with the exception of PDSS1 c.589 A > G, p.(Lys197Glu), c.1118_1120dup, p.(Val373_Gln374insLeu) and COQ2 c.338_341delinsG, p.(Val113_114delinsGly) which were predicted to be ‘polymorphisms’.
Multiple alignment of orthologues showed variable conservation of the protein across different species (Supplementary Fig. 2). Candidate splice site or splice region variants were rare in the gnomAD dataset and predicted to affect the canonical splice site by Netgene2 or SpliceAI (Table 3).
Haplotype analysis of three unrelated families showed a possible ancestral haplotype spanning about 398 kb flanking the PDSS1 gene (Chr10:26,535,625 (rs187101868) to Chr10:26,933,928 (rs111256658)) in family 3. Family 6 shared the telomeric segment and family 5 shared the centromeric segment with two flanking SNPs shared between families 5 and 3 (Chr10:26,720,339 (rs116424900) to Chr10:26,933,928 (rs111256658)) and no flanking SNPs shared between families 5 and 6 (Supplementary Table 2).
The genome-wide filtering pipeline also identified biallelic rare variants in MAST4 (proband, family 1), SCML4 and ZNF813 (proband, family 10) (Supplementary Tables 3–5). The variants in PDSS1 and COQ4 respectively were considered to be the most likely candidates based on the phenotypic similarity (pigmentary retinal changes have been previously reported in some affected individuals suffering from primary COQ10 deficiency) and lack of biological plausibility for other genes. For the proband from family 2, the virtual gene panel analysis identified variants in retinal genes, namely USH2A c.8555 T > C, p.(Leu2852Ser), USH2A c.14074 G > A, p.(Gly4692Arg), RP1 c.4735 T > G, p.(Leu1579Val) and COL4A1 c.4966 C > A, p.(Arg1656Ser). However, only one USH2A variant, c.4735 T > G, p.(Leu1579Val) was considered possibly associated with the phenotype, the second USH2A variant being classified as benign/likely benign in ClinVar. Thus, this case remained unsolved and underwent further analysis leading to the identification of biallelic PDSS1 variants.
Functional assessment of splice variantsThree candidate splice variants, c.468-25 A > G and c.722-2 A > G in PDSS1, and c.682-7 T > G in COQ5 were functionally investigated. RT-PCR of patient blood derived mRNA using oligonucleotide primers targeting PDSS1 or COQ5 (Supplementary Table 6) followed by agarose gel electrophoresis showed definite altered transcript length and mis-splicing only for the COQ5 c.682-7 T > G variant. A reduced canonical transcript band intensity was observed in the sample from family 5, II:1 with the c.468-25 A > G variant in PDSS1. Subsequent Oxford Nanopore Technologies (ONT) single-molecule sequencing of RT-PCR reaction products revealed complex mis-splicing as a result of all three variants. As a result of PDSS1 c.722-2 A > G disrupting the canonical intron 7 splice acceptor site, the altered splicing events were complete exon 7–8 skipping leading to an in-frame deletion of 72 codons (p.(Ala204_Ala277del)) or the use of an alternate splice acceptor site 14 bp downstream of the canonical acceptor site in exon 8 leading to deletion of 14 bp and a frameshift (p.(Gly241Alafs*6) (Fig. 3). The candidate variant PDSS1 c.468-25 A > G was shown to lead to inclusion of an alternate noncanonical exon (Chr11:26,720,135-26,720,359) in the transcript or transcription of the alternate isoform of PDSS1 (NM_001321979.1) with an alternate start codon in the alternate exon 6. For individual family 5, II:1 ONT sequencing of the PDSS1 transcript enabled us to phase the two alleles on independent reads representing independent transcripts at the position of the missense variant (c.589 A > G). Comparing the read depth of the two alleles at this position on full-length reads (primer to primer, exon 1–11), it was clear that the c.589 A allele (representing the c.468-25 A > G variant allele) was underrepresented (27% vs 70%). The mean read depth for each nucleotide position of the alternate versus canonical exon (Chr11:26,720,135-26,720,217 and Chr11:26,720,218-26,720,359) for each allele (c.589 A and c.589 G) showed that 361/400 (90.25%) of the A allele and 159/1619 (9.79%) of the G allele included the alternate exon meaning that the c.468-25 A > G variant led to cryptic splicing in 90.25% of transcript reads. In six control RNA samples, the inclusion of the alternate exon was between 6.5 and 10.14%.
Fig. 3: Detection of mis-splicing caused by variants in PDSS1 by using ONT sequencing.
a IGV visualisation of ONT single molecule sequencing of Individual family 3, II:2. Reads are aligned to the PDSS1 transcript (PDSS1-201 ENST00000376215.10, NM_014317.5) and reads are grouped according to position c.589 A > G. Variant c.722-2 A > G leads to skipping of 222 bp (exon 7–8) or alternate acceptor usage in exon 8 (14 bp downstream). b IGV visualisation of ONT single molecule sequencing of individual from family 5, II:1. Reads are aligned to the human genome (build GRCh38) and PDSS1 exon 6 is shown. Reads are grouped according to position c.589 A > G. Inclusion of the alternate exon (NM_001321979.1) is enriched on the c.468-25 A > G allele (90.25% versus 6.5-10.14% controls). c Schematic representation of cryptic splicing caused by c.722-2 A > G and c.468-25 A > G PDSS1 variants and agarose gel analysis of RT-PCR reactions showing two transcripts in all samples. The 941 bp band corresponds to the canonical transcript and the 1025 bp band corresponds to the alternate exon 6 transcript. PDSS1 NC shows a brighter band for the canonical transcript, while PDSS1 -25 shows a reduced intensity. PDSS1 -2 shows no additional transcripts. PDSS1 NC – normal control, PDSS1 -2 – individual with c.722-2 A > G. PDSS1 -25 – individual with c.468-25 A > G.
One individual carried biallelic COQ5 variants: c.682-7 T > G and c.933delC. One variant is predicted to disrupt splicing and the second predicted to be null. ONT sequencing of the full COQ5 transcript from whole blood of individual family 11, II:1 showed skipping of exon 5 to be the predominant effect of the c.682-7 T > G variant leading to early termination and probable nonsense-mediated decay (p.(Gln230*)) (Fig. 4). In addition, exon 4–5 skipping was identified in a low proportion of reads (3%) leading to a reading-frame shift and early termination (p.(Leu193Phefs*27)). Genotyping of an additional observed SNV (Chr12:120,954,490 C > T) in the unaffected mother demonstrated this was in cis with c.682-7 T > G. This enabled phasing of the two transcript alleles showing that 56% of reads from the c.682-7 G allele showed exon 5 skippings compared to 4% of the trans allele. This suggests that a proportion of transcript is correctly spliced and may lead to mature protein, therefore, suggesting this variant is not a complete LOF allele. A low level of mis-spicing was observed on the trans allele (4%) and in control samples (data not shown) suggesting there may be a basal level of alternate splicing at this position leading to non-expressed transcripts but possibly also a low level of miscalling at the Chr12:120,954,490 position in our data leading to a low proportion of incorrect phasing.
Fig. 4: Detection of mis-splicing caused by c.682-7 T > G in COQ5 by using ONT sequencing.
a IGV visualisation of ONT single molecule sequencing of Individual family 11, II:1. Reads are aligned to the COQ5 transcript (COQ5-201 ENST00000288532.11, NM_032314.4) and reads are grouped according to position c.933delC. Variant c.682-7 T > G leads to skipping of 89 bp (exon 5) and 196 bp (exon 4-5). b Split BAM files according to SNV at Chr12:120,954,490 to show the 56% coverage drop of exon 5 and low level exons 4–5 skipping on the c.682-7 T > G allele compared to low level of exon 5 skipping on the trans allele. c Schematic representation of cryptic splicing events caused by the c.682-7 T > G COQ5 variant and agarose gel analysis of RT-PCR reactions showing alternate splicing. *Variant call observed in both alleles at low quality and low level absent from GS data suggesting an artefact.
Biochemical studiesHuman plasma CoQ10 levels in 8 affected individuals and 3 heterozygotes were within the normal range (reference range 227- 1432 nmol/L, Supplementary Table 7). Individual family 7, II:1 carrying biallelic variants in COQ2 in association with syndromic disease had severely decreased plasma CoQ10 concentration of 39.33 nmol/L.
留言 (0)