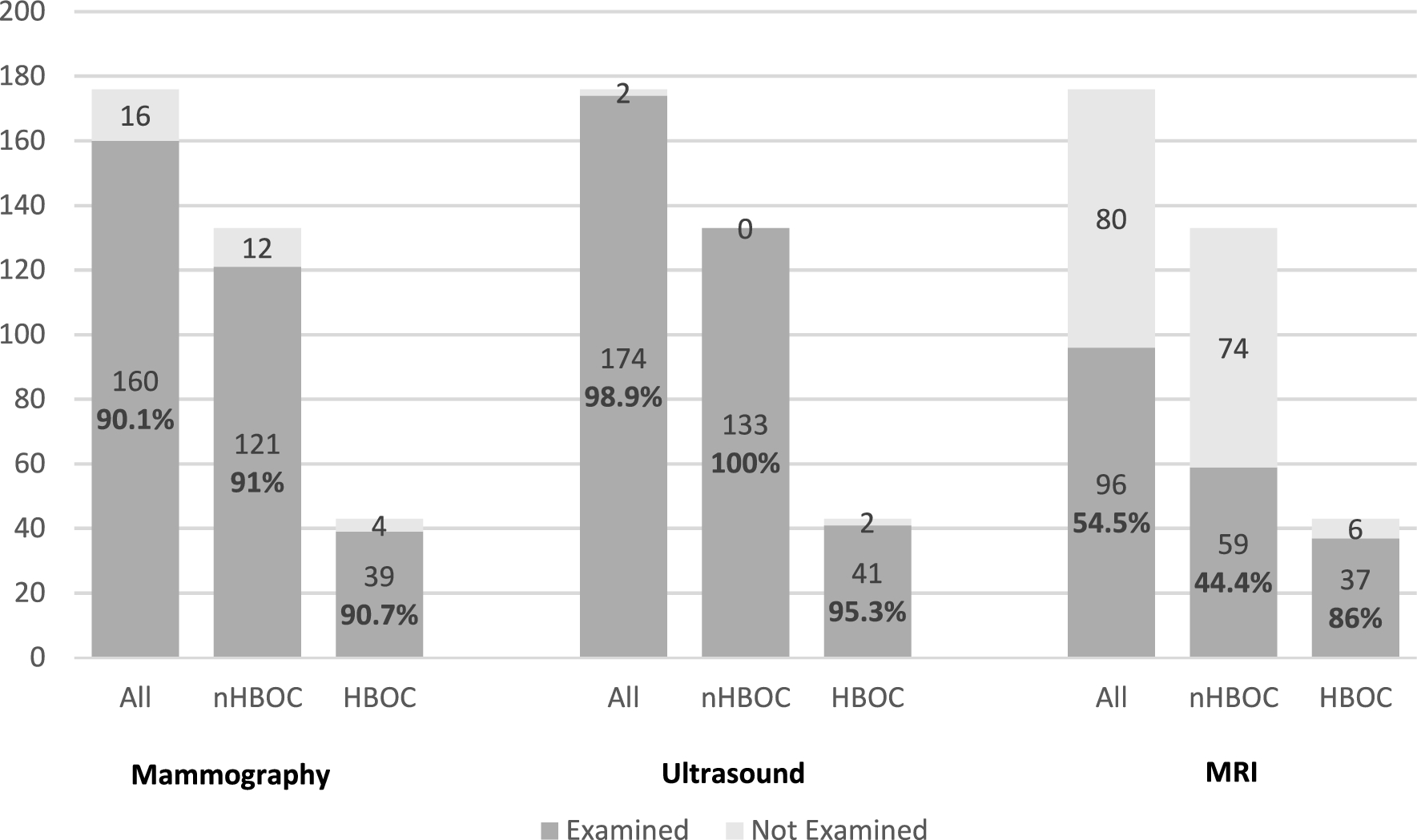
Experimental design
We used the human hepatocellular carcinoma cell line (HepG2) as a study model.
Control group was left untreated. The experimental groups were subjected to treatment with Hepatocyte growth factor (HGF) or chloroquine (CQ) or the combination of HGF and CQ (HCQ) for 24 h. HGF is known to be a potent hepatocyte mitogen, and it was used here as a positive control.
Cell density and morphology as well as the rate of proliferating, respectively, apoptotic cells were investigated using a cytoblock technique. Cell number (CCK-8) and ATP content were investigated using spectrophotometric assays. Lipid accumulation was detected by Oil Red O staining and Triglyceride quantification assay. Autophagy-related proteins (LC3B, p62) and hepatocyte proliferation-related protein (S6K1) were examined using western blot. Autophagic flux was detected by mRFP-EGFP-LC3 plasmid-transfection assay. Each experiment was repeated three times.
Materials
Chemicals and solutions were mainly purchased from Sigma-Aldrich and ThermoFisher. E.g., Hepatocyte growth factor (Sigma-Aldrich, #H9661), Chloroquine (Sigma-Aldrich, #C6628), Fetal Bovine Serum (FBS) (Sigma-Aldrich, #7524), Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, #41965-039) and Penicillin/Streptomycin (C.C.Pro, Z-13-M).
Cell culture
We obtained the HepG2 cells from a previous cooperation partner of the lab (Dr. Kaufmann). HepG2 cells were cultured in 75cm2 cell culture flasks (Greiner bio-one, #108759) with DMEM supplemented with 10% FBS and 1% penicillin and streptomycin (P/S). All cell cultures were maintained at 37 °C in a cell incubator with 5% CO2 and 95% air. A hemocytometer was used for cell counting. HepG2 cells were treated with complete DMEM (control) or with complete DMEM + 40 ng/ml Hepatocyte growth factor (HGF), or with complete DMEM + 60 μM Chloroquine (CQ) or with complete DMEM + 40 ng/ml HGF + 60 μM CQ for 24 h.
Cell counting kit-8 assay
The cell number was determined using a cell counting kit 8 (Abcam, #ab228554). Following the instruction of the manufacturer, HepG2 cells (5 × 103 cells/well) were cultured for 24 h in triplicates using a 96-well tissue culture plate with 100 μL/well of complete DMEM or medium containing 40 ng/ml HGF, 60 μM CQ and 40 ng/ml HGF + 60 μM CQ, respectively. Next, 10 µl of WST-8 solution was added to each well and incubated for another 4 h at 37 °C in the dark. The optical density was read immediately using a spectrophotometric microtiter plate reader (Synergy LX) set at a wavelength of 460 nm.
Immunohistochemistry
The cell proliferation and apoptosis were detected by immunohistochemistry. The cytoblock technique was used for histology to assess cell number and morphology as well as for immunohistochemistry to determine the rate of cells undergoing proliferation or apoptosis using antibodies targeting the Ki-67, respectively, Caspase-3.
At the end of the cell culture observation time, cells were trypsinized and subjected to alcoholic fixation into CytoRich Red solution for at least 24 h. Cells were centrifuged and the pellet was embedded in a gel, followed by paraffin embedding. Sections of 4 µm thickness were prepared. Antigen retrieval was performed according to the requirements of the antibody specification. The sections were incubated with peroxidase blocking solution (Dako, #SM801) for 5 min to quench endogenous peroxidase activity. Primary antibodies mouse anti-Ki-67 antigen clone MIB-1 (Ki-67; Ready to use, Dako, #IR626) and rabbit anti-Caspase-3 cleaved (Caspase-3; 1:50, Zytomed Systems, #RBK009-05) were applied to the sections and were incubated at room temperature (RT) for 20 (Ki-67)/30 (cleaved Caspase-3) minutes. After washing the sections using washing buffer, the second antibody (Dako, #SM802) was applied. The sections were incubated at RT for 20 min. After washing the sections using washing buffer, DAB solution (Dako, #SM803) was applied to the sections and incubated at RT for 10 min. After washing the sections using washing buffer, hematoxylin was applied to the sections followed by 5 min incubation at RT. Sections were scanned by the Hamamatsu Slide Scanner (Nanozoomer XR). Quantitative analysis of sections using HistoCAD VirtualLiver (Fraunhofer Mevis, Bremen, Germany). The HistoCAD VirtualLiver used machine learning techniques to recognize cell positive and negative patterns.
Giemsa staining
The cell morphological changes were detected by Giemsa staining. HepG2 cells were fixed in CytoRich Red solution for at least 24 h. Sections of 4 µm thickness were prepared. Sections were first stained with Giemsa solution at RT for 2 h. After washing the sections using distilled water, acetic acid solution was applied to the sections and incubated at RT for 1 min. Sections were scanned by the Hamamatsu Slide Scanner (Nanozoomer XR).
Hematoxylin–eosin staining
The routine histological examination was performed by Hematoxylin–eosin (H&E) staining. HepG2 cells were fixed in CytoRich Red solution for at least 24 h. Sections of 4 µm thickness were prepared. Sections were first stained with hematoxylin at RT for 5 min. After washing the sections using distilled water, eosin was applied to the sections and incubated at RT for 1 min. Sections were scanned by the Hamamatsu Slide Scanner (Nanozoomer XR).
Oil red O staining
The lipid staining was performed using an Oil Red O staining kit (Sigma-Aldrich, #MAK-194). HepG2 cells (5 × 104 cells/well) were plated triplicates in a 24-well plate with 500 μL/well of complete DMEM. Cells were treated with complete DMEM or medium containing 40 ng/ml HGF, 60 μM CQ and 40 ng/ml HGF + 60 μM CQ, respectively, for 24 h. Then, cells were fixed in 10% formalin for 1 h, the formalin was discarded and the cells were washed three times with ultrapure water. The cells were incubated with 60% isopropanol for 5 min. The 60% isopropanol was discarded and the cells were incubated with Oil Red O Working Solution for 20 min. The Oil Red O Working Solution was discarded and the cells were washed three times with ultrapure water. Next, hematoxylin was added to the cells and incubated for 1 min. The hematoxylin was discarded and the cells were washed three times with ultrapure water. Cells were covered with 500 µl ultrapure water. Visualization of Oil Red O staining in HepG2 cells was performed using a Leica optical microscope (DM IRB).
Triglyceride quantification assay
The cellular triglyceride (TG) content was measured using a triglyceride quantification kit (Abcam, #ab65336). Triglyceride standard curves were established using the triglyceride standards provided in the kit. 1 × 107 cells were harvested and lysed to prepare cells lysates. 50 μl reaction mixture was prepared for each reaction. The triglyceride standard wells (50 μL/well), sample wells (50 μL/well) and sample background control wells (50 μL/well) were set up in a 96-well tissue culture plate. 2 μL of lipase was added to the standard wells and sample wells, 2 μL of triglyceride assay buffer was added to the sample background control wells and incubated at RT in the dark for 20 min. 50 μl reaction mixture was added to each standard well, sample well and sample background control well and incubated at RT darkroom for 60 min. The output optical density was read immediately using a microplate reader (Synergy LX) at the wavelength of 570 nm.
ATP detection assay
The ATP assay was performed using an ATP detection kit (Abcam, #ab83355). ATP standard curves were established using the ATP standards provided in the kit. 3 × 106 cells were harvested and lysed to prepare cells lysates. Deproteinizing Sample Preparation Kit (Abcam, #ab204708) was used to remove superfluous enzymes. 50 μl reaction mixture and background mixture were prepared for each reaction. The ATP standard wells (50 μL/well), sample wells (50 μL/well) and sample background control wells (50 μL/well) were set up in the 96-well tissue culture plate. 50 μl reaction mixture was added to each standard well and sample well, 50 μL background reaction mixture was added to each sample background control well and incubated at RT in the dark for 30 min. The optical density was read immediately using a microplate reader (Synergy LX) at the wavelength of 570 nm.
Western blotting
Cells were lysed in the RIPA buffer (Sigma, #R0278) containing a protease and phosphatase inhibitor cocktail (Thermo Scientific, #78442). The concentration of total proteins was measured using the BCA protein assay kit (Thermo Scientific, #23225) and a Synergy LX Multi-Mode Microplate Reader. Equal amounts (20 µg) of protein were denatured with Laemmli sample buffer. Proteins were separated in electrophoresis and transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were washed with Tris-buffered saline with tween 20 (TBST) and blocked with 5% BSA-TBST buffer. Primary antibodies rabbit anti-light chain 3 (LC3; 1:1000, Abcam, #ab48394), rabbit anti-SQSTM1 (p62; 1:10,000, Abcam, #ab109012), rabbit anti-S6K1 (1:2000, Abcam, #ab32359) and rabbit anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1:15,000, Abcam, #ab181602) were applied to the membranes and were incubated at 4 °C overnight. After washing the membranes using TBST, the second antibody (polyclonal goat antibody to rabbit IgG; 1:10,000, Abcam, #ab6721) was applied. The PVDF membranes were incubated at 37 °C for 1 h. After being probed using enhanced chemiluminescence western blotting detection reagents (GE Healthcare, #RPN2209), signals were visualized using Fusion FX7 (Labtech International Ltd, Heathfield, United Kingdom). Quantitative analysis of western blotting results was performed using the software Image J.
mRFP-EGFP-LC3 plasmid transfection
The HepG2 cells were transfected with the mRFP-EGFP-LC3 (ptfLC3) plasmid (Addgene plasmid, #21074, ptfLC3 was a gift from Prof. Tamotsu Yoshimori) using Lipofectamine 2000 kit (Lipofectamine, #11668019) according to the instruction (Kimura et al. 2007). Briefly, we isolated a single colony of bacteria from the Lysogeny broth (LB) agar plate, followed by placing the colony into the LB Broth medium (Sigma, #L2542) for further expansion. Next, a plasmid DNA extraction kit (QIAGEN Plasmid Midi Kit, #12143) was used to isolate the plasmid DNA. Plasmid DNA was transfected into HepG2 cells using the Lipofectamine 2000 kit. 24 h after transfection, cells were cultured in complete medium or medium containing 40 ng/ml HGF, 60 μM CQ and 40 ng/ml HGF + 60 μM CQ for 24 h, respectively, at 37 °C in a cell incubator with 5% CO2 and 95% air. Visualization of transfected HepG2 cells was performed using a Zeiss fluorescence microscope (AX10).
Statistical analysis
Data analysis was performed with the statistical software SPSS 25.0 and SigmaPlot 13.0 (Statcon, Witzenhausen, Germany). The differences between groups were assessed using the one-way ANOVA test with LSD post hoc analysis (Data follow normal distribution or approximately normal distribution and homogeneity of variance) or Kruskal–Wallis test (Data do not follow normal distribution or homogeneity of variance). We pooled the data from three rounds of repeated experiments for statistical analysis. Statistical differences were considered significant when P < 0.05.




留言 (0)