
記住我
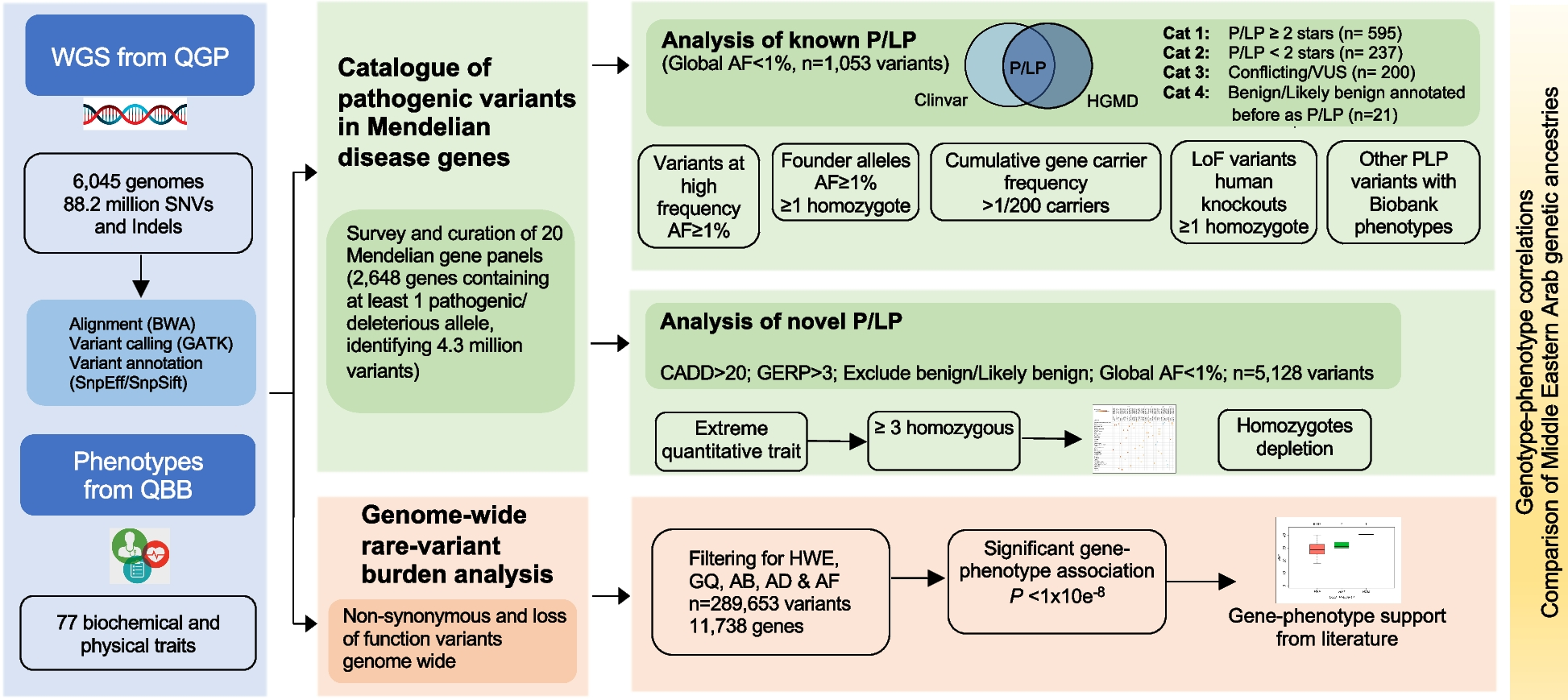
ClinVar database files clinvar_20190102.vcf.gz and clinvar_20200609.vcf.gz were downloaded from https://www.ncbi.nlm.nih.gov/clinvar/ [23] under the GRCh38/hg38 genome assembly. Variants in the older file were used in the training phase of model development. Next, we prepared separate training sets for point variants and insertion/deletions (InDels). For SNVs, nonsynonymous SNVs (nsSNVs) labeled “Pathogenic” or “Likely pathogenic” were used as true positives (TPs), and nsSNVs labeled “Benign” or “Likely benign” were used as true negatives (TNs). Variants with conflicting clinical interpretations were removed. Conflicting clinical interpretations were defined as one of these scenarios: conflict between benign/likely benign and variants of unknown significance (VUS), conflict between pathogenic/likely pathogenic and VUS, or conflict between benign/likely benign and pathogenic/likely pathogenic. Variants that were absent from at least one of the three datasets (gnomAD [21], ExAC [20], and the 1000 Genomes Project [19]) were retained. A further filter removed any nsSNVs that were absent in all three datasets. We consider this to be a good trade-off between preserving important allele frequency information and removing “easy-to-classify” variants during training. In the end, 26,517 rare nsSNVs with 9009 TPs and 17,508 TNs (Additional file 1: Table S1) were used for training. For InDels, the same criteria were applied to obtain TPs and TNs. Additionally, only InDels annotated as non-frameshift (nfINDELs) and having lengths >1 and ≤ 48 base pairs were included. A total of 1981 rare nfINDELs with 1306 TPs and 675 TNs passed the filtering criteria and were used to train the MetaRNN-indel model (https://github.com/Chang-Li2019/MetaRNN) [29] (Additional file 1: Table S2).
Test setsWe constructed 7 test sets to evaluate the performance of our SNV-based model, namely, MetaRNN (https://github.com/Chang-Li2019/MetaRNN) [29] (summary in Additional file 1: Table S3) with 24 other methods, including MutationTaster [10], FATHMM [30], FATHMM-XF [12], VEST4 [9], MetaSVM [31], MetaLR [31], M-CAP [17], REVEL [4], MutPred [16], MVP [8], PrimateAI [15], DEOGEN2 [14], BayesDel_addAF [7], ClinPred [6], LIST-S2 [5], CADD [3], Eigen [13], GERP [32], phyloP100way_vertebrate [33], phyloP30way_mammalian, phyloP17way_primate, phastCons100way_vertebrate, phastCons30way_mammalian, and phastCons17way_primate. The first test set (rare nsSNV test set, RNTS) was constructed from rare pathogenic nsSNVs with a maximum population allele frequency (AF) of 0.01 that were added to the ClinVar database after 20190102 and rare nsSNVs with a maximum population AF of 0.01 that were present in all three population datasets while not reported in ClinVar and matching on genomic location (randomly selected non-pathogenic nsSNVs within 10 kb from the pathogenic ones), resulting in 11,540 variants with 5770 TPs and 5770 TNs (Additional file 1: Table S4). The second test set (rare clinvar-only test set, RCTS) was constructed from recently curated (after 20190102) ClinVar rare pathogenic nsSNVs (n = 6190) and rare benign nsSNVs defined as having a maximum AF<0.01 in all population datasets (n = 11,811) (Additional file 1: Table S5). The third test set (de novo RCTS, DN-RCTS) was constructed from RCTS with 0 AF in all population datasets, which resulted in 4537 TPs and 831 TNs (Additional file 1: Table S6). The fourth test set (all allele frequency set, AAFS) was constructed from all pathogenic and benign nsSNVs added to the ClinVar database after 20190102 regardless of AF, resulting in 29,924 variants with 6205 TPs and 22,808 TNs (Additional file 1: Table S7). The fifth test set, the TP53 test set (TP53TS), was constructed from the TP53 mutation website (https://p53.fr/index.php) [34]. Variants with median activity <50 were considered pathogenic, while variants with median activity ≥100 were considered benign. After removing variants used in the training set, 824 variants remained with 385 TPs and 439 TNs (Additional file 1: Table S8). The sixth test set was retrieved from a recent publication (Additional file 1: Table S9) [26]. The TPs (n = 878) were curated from cancer somatic variant hotspots, and the TNs (n = 1756) were curated from the population sequencing study DiscovEHR [35]. The Human Gene Mutation Database (HGMD) (https://www.hgmd.cf.ac.uk/) [24] is another popular database that provides high-quality disease-associated variants. As the last test set, we retrieved all DM variants (disease mutation; the class of variants in HGMD with the highest confidence of being pathogenic) from HGMD Professional version 2021.01. Only variants reported in dbNSFP (https://sites.google.com/site/jpopgen/dbNSFP) [27, 28] as missense were kept. We further removed variants that were reported in the HGMD Professional version 2017 to avoid unfair comparisons with scores that used HGMD in their training process. Additionally, variants reported in ClinVar 20200609 as pathogenic, likely pathogenic, benign, or likely benign were filtered out to explore the generalizability of our score to independently curated disease-causing variants. These filtered nsSNVs were used as TPs. For true negatives (TNs), we used rare nsSNVs that were observed in gnomAD v3 with allele frequencies between 0.01 and 0.0001 as a trade-off between their rarity and probability of being truly benign. The number of TP and TN variants were matched using random selection, which resulted in 45,256 nsSNVs in total (22,628 TP variants and 22,628 TN variants). For our InDel-based model, namely, MetaRNN-indel, the first test set was constructed from InDels added to the ClinVar database after 20190102, which resulted in 828 InDels with 365 TPs and 463 TNs (Additional file 1: Table S10). The second test set was constructed from HGMD Professional version 2021.01. All the nfINDELs that were not used in training MetaRNN-indel were kept as TP. For TN, rare nfINDELs with AF less than 0.01 were retrieved from gnomAD v2.1.1 as TNs, which were then randomly sampled to match the number of TPs. A total of 8020 nfINDELs (4010 TP variants and 4010 TN variants) were collected after filtering.
Flanking nsSNVsAfter obtaining all data sets of target variants, we retrieved nsSNVs from their flanking sequences using dbNSFP4.1a [27, 28]. Specifically, the genomic location of the variant and the affected amino acid position of the protein and the affected codon were first identified in dbNSFP. Then, a window size of ± 1 codon around the affected codon was identified, and all nsSNVs inside this window were retrieved with a maximum length of 9 base pairs (bps). For a given target variant, the maximum possible number of nucleotides on either side is 5 bps (3 bps from one flanking codon and 2 bps from the target codon). To center the input window on the target variant and have a uniform shape for all inputs, we padded the input window to reach an 11-bp window for each target variant so that there were 5 bps on each side of the target variant. This window, including the target variants, was used as one input for our model (Fig. 1).
Fig. 1
Data preparation and model development steps for MetaRNN. First, the target variant and affect codon were identified, e.g., g.1022225G>T. Second, the flanking sequences were retrieved as well as all possible alternative alleles, as illustrated on the nucleotides to the right of the up-pointing arrow. Third, only alleles that result in a missense variant were kept, and annotation scores were averaged across these alleles within the same locus. For example, the annotation scores for variants g.1022230A>T and g.1022230A>C will be averaged, to get the locus-specific annotations. Lastly, the model will be trained using the annotations for context variants and annotations for the target variant
For each position, multiple alternative alleles may exist. For each annotation at context loci, we calculated the average score across all alleles that would result in a nonsynonymous variant at the locus. This averaged annotation score was then used to represent the locus for that annotation. This setup has the advantage of keeping the most critical context information while limiting the unnecessary noise introduced by having inconsistent order and dimension of alleles at different loci, e.g., some loci may possess three nsSNVs while others may include only one nsSNV. The target variant would directly use annotation scores for the observed allele. We assume that these nsSNVs and related annotations can capture the most critical context information concerning the pathogenicity and functional importance of the amino acids. We assumed that context nsSNVs provided all the critical information, so we ignored any synonymous variants in composing the context information. After these steps, the input dimension for the MetaRNN model becomes 11 (bps) by 28 (features, see below).
The same rules to adopt the flanking region were applied to InDels with one difference: instead of affecting only one codon, target InDels may directly affect multiple codons simultaneously. Thus, the ± 1 codon window was defined as the window beyond all the directly affected codons. For deletions, target variants were those loci deleted by the variant, and their annotations were averaged for each locus. For insertions, since no annotation is available for the inserted nucleotides, we used annotations from loci adjacent to the insertion position as surrogates. Since we focus on short InDels with length ≤ 48, with 5 bps around each side as context information, the input dimension for the MetaRNN-indel model is 58 (bps) by 28 (features, see below). Again, the synonymous variants were ignored in composing the context information.
Feature selectionFor each variant, including target nsSNV/nfINDEL and flanking region nsSNVs, 28 features were either calculated or retrieved from the dbNSFP database, including 16 functional prediction scores: SIFT [36], Polyphen2_HDIV [37], Polyphen2_HVAR, MutationAssessor [11], PROVEAN [38], VEST4 [9], M-CAP [17], REVEL [4], MutPred [16], MVP [8], PrimateAI [15], DEOGEN2 [14], CADD [3], fathmm-XF [12], Eigen [13], and GenoCanyon [39]; eight conservation scores including GERP [40], phyloP100way_vertebrate [33], phyloP30way_mammalian, phyloP17way_primate, phastCons100way_vertebrate, phastCons30way_mammalian, phastCons17way_primate, and SiPhy [41]; and four calculated allele frequency (AF)-related scores. The highest AF values across subpopulations of the four data sets from three studies, namely, the 1000 Genomes Project (1000GP), ExAC, gnomAD exomes, and gnomAD genomes, were used as the AF scores. All missing scores in the dbNSFP database were first imputed using BPCAfill (http://ishiilab.jp/member/oba/tools/BPCAFill.html) [42], and all scores were standardized before feeding to the model for training. Some more recently developed scores were excluded to minimize type I circularity in training our ensemble model, including MPC and ClinPred, which used ClinVar variants in their training process.
Model developmentWe applied a recurrent neural network with gated recurrent units [22] (GRU) to extract and learn the context information around target variants (Fig. 1). Bayesian Hyperparameter Optimization [43] was used to determine the best-performing model structure from a wide range of model structures. Specifically, the input layer takes an 11 × 28 matrix as input for the MetaRNN and a 58 × 28 matrix for the MetaRNN-indel model. After the bidirectional GRU layer, the MetaRNN model cropped out the context information, and only the learned features for the target variant were kept. This setup can significantly reduce the number of parameters compared to keeping all context features in the subsequent dense layer. Following the same idea, for MetaRNN-indel, the output for the last bidirectional GRU layer only returns the prediction for the final locus (return_sequences parameter was set to false) to limit the number of possible parameters in the following dense layer. The output layer is composed of 1 neuron with a sigmoid activation to model our binary classification problem. A binary cross-entropy loss was used as the loss function, and the Adam optimizer [44] was used to update model parameters through backpropagation [45]. This process used 70% of the training data for model training and 30% of the training data for performance evaluation, so no test sets were used in this step. The Python packages sci-kit-learn (https://scikit-learn.org/stable/) [46] and TensorFlow 2.0 (https://www.TensorFlow.org/) [47] were used to develop the models, and KerasTuner (https://keras-team.github.io/keras-tuner/) [48] was adopted to apply Bayesian Hyperparameter Optimization. The search space for all the hyperparameters is shown in Additional file 1: Table S11. The models with the smallest validation log loss were used as our final models for nsSNV (MetaRNN) and nfINDEL (MetaRNN-indel).
Comparison of the performance of individual predictorsAs a model diagnosis step, SHAP (SHapley Additive exPlanations) values were calculated to measure each feature’s contribution to the predicted consequence of variants [49]. We first used 100 random samples from our training data to calculate the background distribution of the values. Next, feature permutations were performed using 100 random samples from our validation data (RNTS). Since the variance of the estimates scale by 1/sqrt(background sample size), we chose to use 100 samples, which would give a reasonable estimate. The Python library SHAP (https://shap.readthedocs.io/en/latest/index.html) [49] was used to calculate SHAP values and plot visualizations.
To quantitatively evaluate model performance, we retrieved 39 prediction scores from dbNSFP to compare with our MetaRNN model including MutationTaster [10], MutationAssessor [11], FATHMM [30], FATHMM-MKL [50], FATHMM-XF [12], PROVEAN [38], VEST4 [9], MetaSVM [31], MetaLR [31], M-CAP [17], MPC [18], REVEL [4], MutPred [16], MVP [8], PrimateAI [15], DEOGEN2 [14], BayesDel (AF and noAF models) [7], ClinPred [6], LIST-S2 [5], LRT [51], CADD (raw and hg19 models) [3], DANN [52], Eigen (raw and PC models) [13], GERP [32], Polyphen2 (HVAR and HDIV) [53], SIFT4G [54], SiPhy [41], GenoCanyon [55], fitCons (integrated) [56], phyloP (100way_vertebrate, 30way_mammalian and 17way_primate) [33], and phastCons (100way_vertebrate, 30way_mammalian and 17way_primate) [57]. The corresponding rank scores were retrieved for each of these 39 annotation scores to facilitate comparison. For the MetaRNN-indel model, four popular methods were compared, including DDIG-in (http://sparks-lab.org/server/ddig/) [58], CADD (https://cadd.gs.washington.edu/) [3], PROVEAN (https://www.jcvi.org/research/provean) [38], and VEST4 (http://cravat.us/CRAVAT/) [59]. For the ClinVar holdout test set, all four methods were compared with MetaRNN-indel. For the HGMD test set, VEST4 was removed from the comparison since it used HGMD InDels during training, and we did not have access to an older version of HGMD with InDels to exclude these variants. For both test data sets, LiftOver was used to convert hg38 genomic coordinates to GRCh37/hg19 for DDIG-in and PROVEAN. DDIG-in scores were retrieved from https://sparks-lab.org/server/ddig/ [58]. VEST4 indel scores were retrieved from http://cravat.us/CRAVAT/ [59]. The CRAVAT format was used, and each InDel variant was assumed to be located on both + and – strands. For PROVEAN indel, the scores were retrieved from http://provean.jcvi.org/genome_submit_2.php?species=human [
留言 (0)