
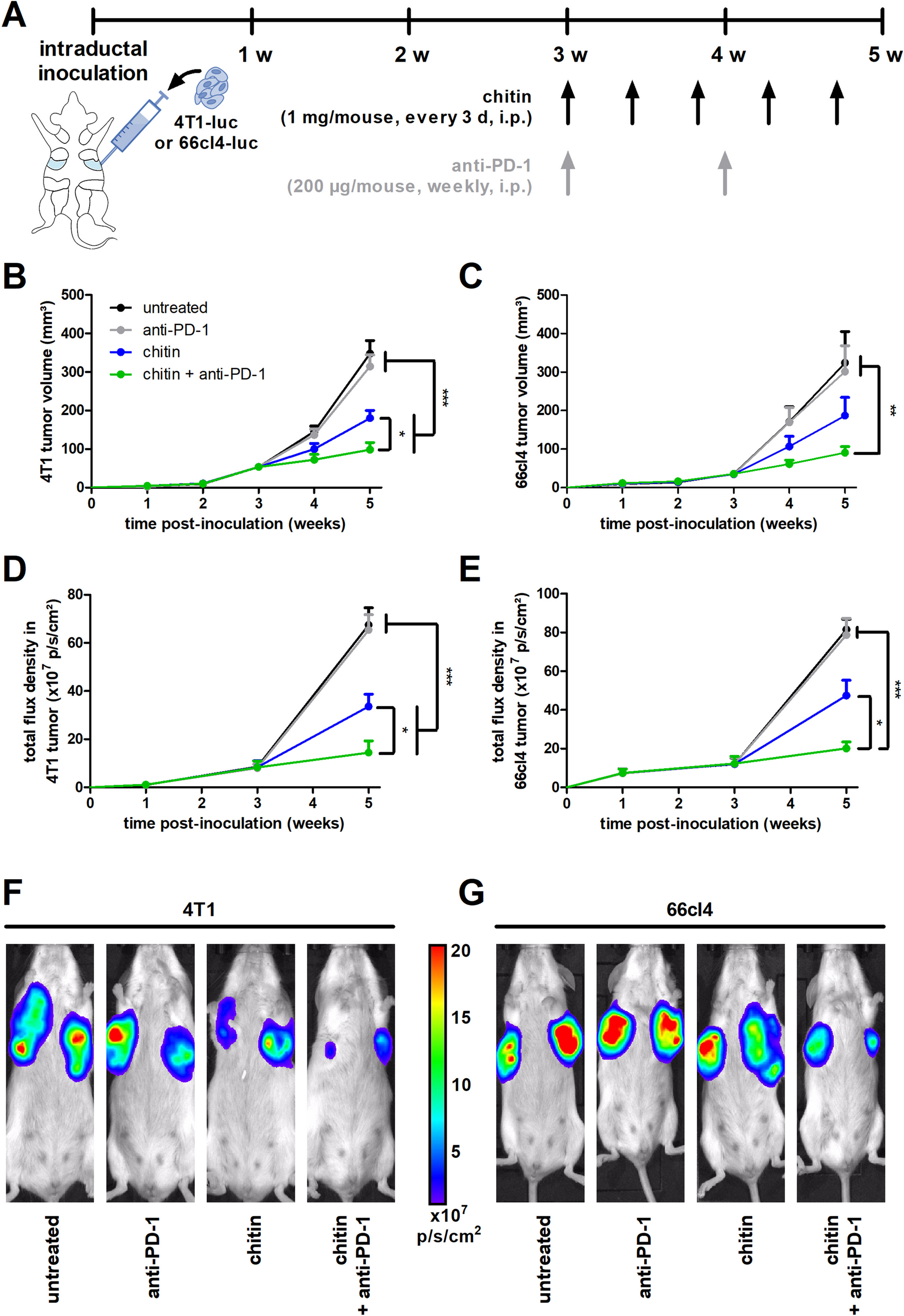
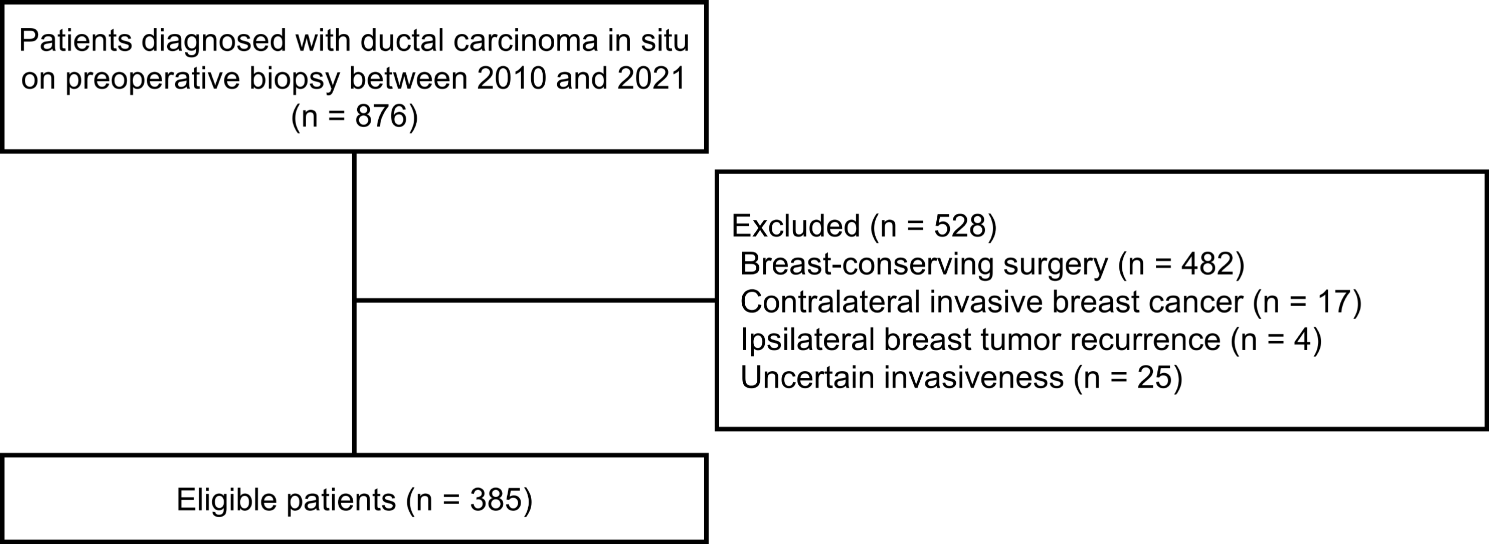
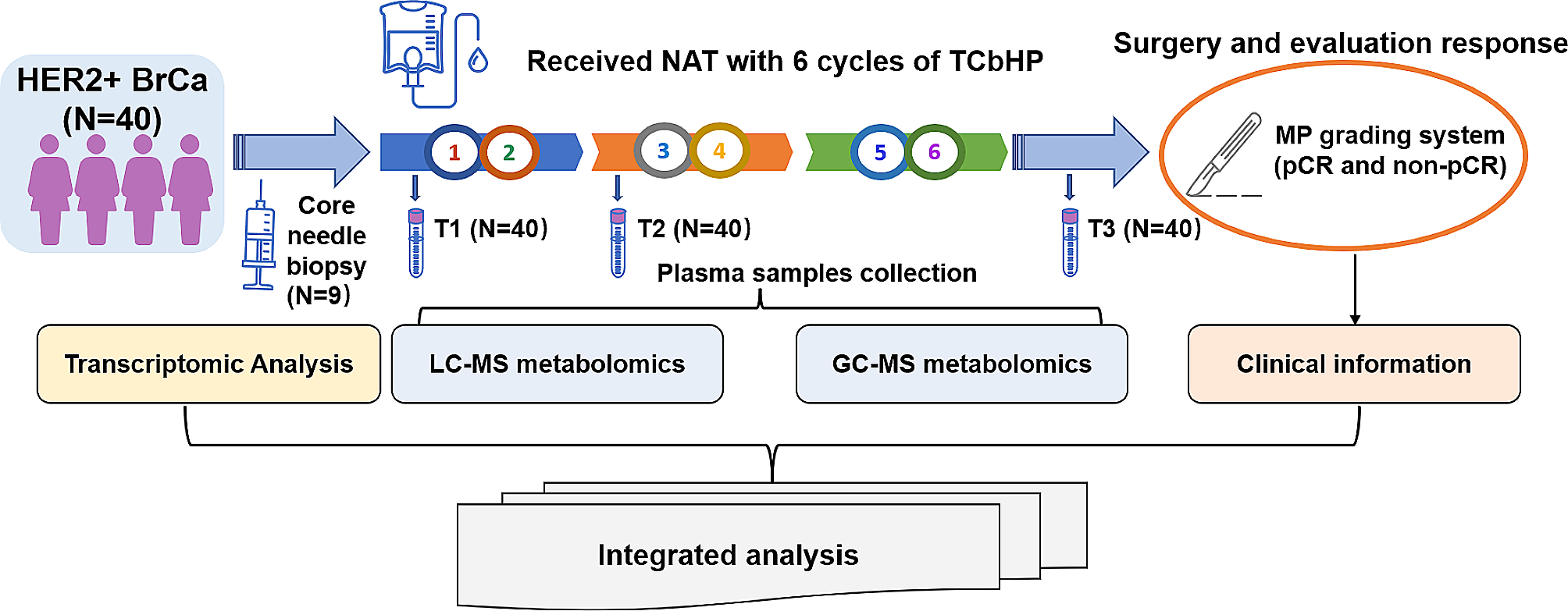
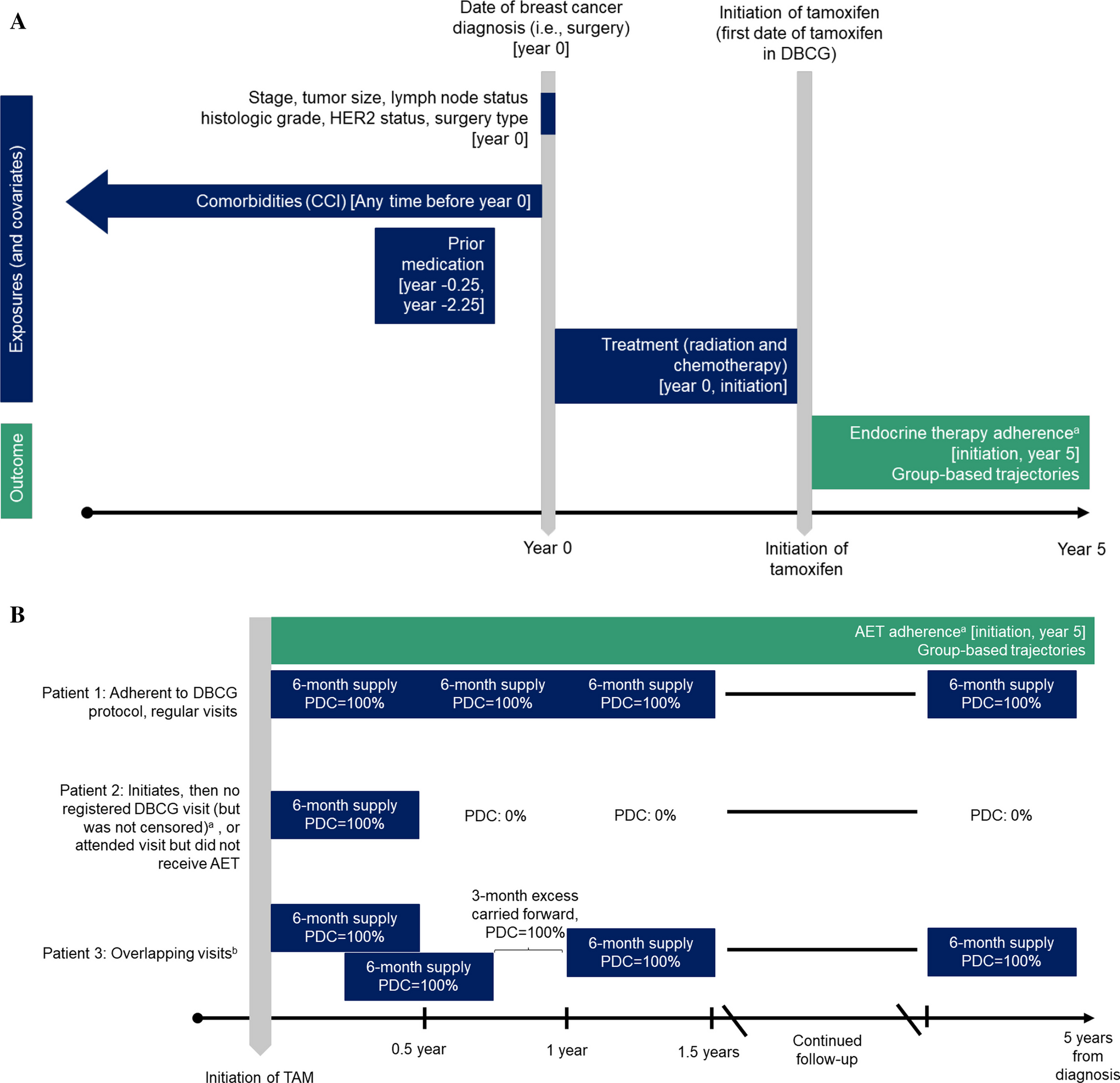
記住我
According to a PubMed search, the top six studied BCCLs are MCF7, MDAMB231, T47D, SKBR3, MCF10A and MDAMB468, which cover more than 90% of all BCCL associated studies across more than 90,000 publications (Additional file 1: Fig. S1). Previous studies on these cell lines are restricted to whole-exome or low-pass whole genome sequencing at 0.2X coverage [5]. Here we describe whole genome sequencing (WGS) of these models on the Illumina X10 platform with an average ~ 60 × coverage, including two replicates of the most-commonly used MCF7 cell line (~ 53× and ~ 78×). Raw reads were mapped to human genome GRCh37 and single nucleotide variants were called using the Issac pipeline [22]. WGS is the most accurate way to assess cell line identity [26]. We have compared the genotyping calls from our WGS data with SNP array from two independent studies previously published [3, 12]. Correlation analysis of the genotyping calls at the same genomic loci shows high concordance between ours and the published data (R > 0.9) (Additional file 1: Fig. S2), confirming the identity of all cell lines used in this study. In total, we have identified 3,540,312 to 4,108,844 variants per sample from the WGS data, including SNVs and small indels (Additional file 2: Table S1). The majority of variants are present in the dbSNP database [25] (94.0–96.2%) (Additional file 2: Table S1). About 90% of these dbSNP variants have also been reported in the 1000 genome projects [10], suggesting most variants in these cell lines models represents common variants in the human population, similar to the previous finding of WGS of the Hela cell line [15]. However, each of the cell lines has about 5% of total variants that are cell line specific variants (Fig. 1A).
Fig. 1
Summary of single nucleotide variations (SNVs) in six breast cancer cell lines, including two replicates for MCF-7. A Classification of SNVs into different categories: overlap with 1000 genome project; overlap with dbSNP but not in 1000 genome project; cell-line specific events; B Distribution of the SNVs in MCF-7 in respect to location of protein coding genes. “RNA variant” are the variants that lie on one of the RNA transcripts C unsupervised hierarchical clustering of MCF7 in this study together with 27 MCF7 strains in Ben-David et al., based on their missense mutation obtained from supplementary information from (Ben-David et al.) D Percentage of overlapping missense mutations between our findings and those identified in multiple strains of MCF7s in Ben-David et al. E Venn diagram shows the overlap of missense mutations in Ben-David et al. and this study
In MCF7, for example, more than half of the cell line specific variants are located in inter-genic regions and ~ 1/3 are found in introns (Fig. 1B). This is similar across all the cell lines models (Additional file 1: Fig. S3). We compared the list of missense mutation in MCF7 to a recent study of multiple strains of MCF7s [5] (Fig. 1C). Most of the missense mutations in [5] shared between MCF7 strains, therefore likely to come from the founder tumor, were have been identified in this study (20/23), and the concordance with our data increases as the number of mutant MCF7 strains increases (Fig. 1D, E). In addition to the mutations reported in Ben-David et al., our WGS data identified 9,555 additional missense mutations, 635 of which are not reported in 1000 genome or dbSNP database (Additional file 2: Table S2). Sequencing of large cohorts of breast cancer tissue has revealed recurrent mutations in long non-coding genes including MALAT1 and NEAT1[20]. Among all the cancer associated long non-coding genes reported [21] ([9], Lnc2Cancer v3.0), we identified mutations of MALAT1, HOTAIR and ZFAS1 in these cell line models. For example, Malat1 showed a heterozygous mutation (chr11:65271832T > C) in MCF7, but not in MDA-MB-231 (Additional file 1: Fig. S4). The complete list of variants in the non-coding regions in these cell lines models (Additional file 2: Table S3) could serve as a useful database for selection of models in non-coding RNA studies.
We also analysed the mutational signatures [2] using “deconstructSig” r package in the cell lines [24] (Methods). There is no difference between cell lines, likely due to the high number of mutations (Fig. 2A). As we do not have germline genomic data for cell line donors, we have used missense mutations for the cell lines after filtering against the dbSNP and 1000 genomes databases. Signatures 1A/1B, 3 and 6 are commonly observed across all the cells lines. Signature 1 has previously been commonly observed across all cancer types, while Signature 3 has been found in breast, ovarian and pancreatic cancers (Fig. 2B and Additional file 2: Table S4). Interestingly, Signature 3 is associated with DNA double-strand break-repair and germline and somatic BRCA1 and BRCA2 mutations. Signature 6 has been found in a majority of cancers and is associated with DNA mismatch repair and microsatellite unstable tumours.
Fig. 2
Mutational signature analysis of the Breast Cancer Cell Lines. A Output mutational profiles of the six cells lines from deconstructSigs displaying the fraction of mutations found in each trinucleotide context B pie charts of the mutational signatures identified for each of the six cell lines models from deconstructSigs
In addition to the SNVs, we also called SVs and CNVs from the WGS data using different methods, including Breakdancer [7] and Delly [7] for structural variants; CNVnator [1] and Lumpy [1] for copy number (see Methods). A summary of SVs and CNVs identified in MCF7 and the other four cell lines is shown in Fig. 3A and Additional file 1: Fig. S5 respectively. In total, we have identified 321 inter-chromosomal SVs, with 38–108 SVs per cell line (Additional file 2: Table S5). We use the GREAT program [17] to perform pathway analysis of the SVs events from individual cell lines. As expected, the top enriched pathways are genes in amplified regions previously identified from breast cancer, such as genes like ZNF217 and BCAS3 in MCF7 [11]; a selected list of luminal genes have also been enriched in the luminal cell lines MCF7 and T47D (Additional file 1: Fig. S6).
Fig. 3
Genomics landscape of copy number and structural variants in breast cancer cell lines. A Representative circos plot of MCF7 for genomics alterations. Copy number events are summarized in the inner circle with red and blue colour indicates copy number gains and blue respectively, two inner circles are represented two replicates of MCF7 samples (one 30X and one 60X). Arcs connecting two loci of difference chromosomes indicate inter-chromosomal structural variations. B Venn diagram shows the overlap of CNV genes between COSMIC, Ben-David et al. and this study. C Representative genome browser view of copy number alterations covering a common CNV gene, CDKN2A. Tracks from top to bottom: depth of coverage in an NA12878 control (control), all reads in the sample (all reads), or reads with mapping quality > = 20 (MQ > 20), the average mapping quality of aligned reads from the sample (MQ, if no reads align MQ = 0), coverage standard deviation from 500 controls (Coverage SD, indicating common CNV), overlapping segmental duplications published by Bailey JA et al. 2002 (SEG-DUP, used as control for germline CNVs), discordant pairs (DP), split reads (SR), variants from the Database of Genomic Variants (DGV), and RefSeq genes (Genes). D Representative genome browser view plot of a novel CNV gene LINC00290 in this study
In addition to SVs, we also identified copy number alterations in these cell line models (Additional file 3: Table S6). WGS has improved accuracy in detecting CNVs compared to exome-seq due to its uniform coverage [4]. We compared the CNVs identified from MCF7 with existing data from array-based COSMIC and shallow WGS from Ben-David et al. (Fig. 3B, Additional file 4: Table S7). Ben-David et al. reported that high variability in copy number calling, our study is more concordant with both of the studies than they are with each other (Fig. 3B). For example, a substantial proportion (138/478) of the CNVs identified in this study but not in COSMIC are reported in Ben-David et al. using shallow WGS, suggesting the array-based method missed a lot of CNVs due to poor coverage. WGS can identify the known key copy number events in each of the cell lines with fine resolution, including AURKA and MYC amplification, CDKN2A (Fig. 3C) deletion in MCF7, ERBB2 amplification in SKBR3, and CDKN2A deletion in MDAMD231 (Additional file 3: Table S6). Our WGS data can also identify novel CNVs not reported in either COSMIC or Ben-David et al. LINC00290, a long non-coding RNA, which has been previously reported to undergo copy number loss in a pan-cancer study (Zack et al. 2013), has a homozygous deletion in MCF7 (Fig. 3D). Furthermore, CNV boundaries were accurately detected by WGS compared to WES, in many cases with base pair precision (Additional file 3: Table S6). For example, in MCF7 cell line, the boundaries of the homozygous deletion cover only CDKNA2A, and the nearby CDKN2B is in a hemizygous deletion region (Fig. 3C). FOXA1 copy number gain is in a focal amplicon, whereas GATA3 is in a broad copy number gain region about 0.5 Mb long & ESR1 copy number gain is towards the 5’ end of the gene (Additional file 1: Fig. S7).
In order to estimate the functional impact of these variants, we compared our list of copy number variations with functional screen data from these cell lines [16]. In MCF7, 33 out of the 445 copy number events harbour at least one of the breast cancer essential genes from [16]. Interestingly this reveals coordinated amplification of ESR1 and its co-factors NCOA3 and GATA3 and pioneer factor FOXA1, perhaps explaining the extreme estrogen-sensitivity of this cell line (Additional file 3: Table S6). Therefore, this study resolves known copy number events at nucleotide resolution and reports a substantial number of new copy number variants, some with evidence for function in these commonly used cell line models.
Patient-derived xenograft modelsBruna et al. performed shallow WGS (< 1X) profiling of 83 breast cancer PDX models and assessed their response to drug treatment [6]. Copy number calls were generated at 100 kb resolution. Here we have selected six well-established and frequently used PDX models with two matched blood samples [8] and performed WGS with at least 90 × coverage (blood samples were sequenced at 30 × coverage) (Additional file 5: Table S8) to generate reliable SNP, copy number and structural variant calls. Using the same pipeline as the cell line analysis based on the reads mappable to human genome, we identified 3,435,230 to 4,172,800 variants per sample from the WGS data, including SNVs and small indels (Additional file 2: Table S1). Similarly to the cell line data, the majority have been identified in the dbSNP database [25] (95.6–95.9%) (Additional file 1: Table S1). About 84% of these variants identified in dbSNP have also been reported in 1000 genome projects [10]. Each of the PDXs has about 4% of total variants as PDX specific variants (Fig. 4A). In HCI002, more than half of the PDX line specific variants are located in inter-genic regions, followed by variants in introns (Fig. 4B). While HCI004, HCI008 and two blood samples have the least number of variants in intron regions (Additional file 1: Fig. S8). To validate our findings, we performed exome sequencing analysis of selected models and identified There are 12 non-synonymous mutations identified using an independent exome sequencing analysis of these models, all of which have been also identified in our WGS data (Additional file 5: Table S9). Similar to the cell lines, we also performed mutational signature analysis in the PDX models. For PDX samples, we are able to generate somatic missense mutation lists for the HCI004 and HCI0010 models as we have matched blood samples for these models and can filter sample-specific germline variants in additional to 1000 genomes and dbSNP (Fig. 5A). While Signatures 1A/1B, 3 and 6 are found in most of the PDX models, there are also model-specific signatures (Fig. 5B). Interestingly, Signature 2, which is associated with hypermutation or kataegis, is found in the HCI004 model only. Other model-specific signatures including Signature 5 for HCI008 and Signature 3 for HCI010 and HCI012 (Additional file 5: Table S10).
Fig. 4
Summary of SNVs in breast cancer patient-derived xenograft models: A classifications of SNVs into three categories: overlap with 1000 genome project; overlap with dbSNP but not in 1000 genome project; PDX-specific variants. B Distribution of the SNVs in HCI002 model in respect to location of protein coding genes. C Representative genome browser view of copy number alteration covering PTEN in HCI004 model. Tracks from top to bottom: depth of coverage in HCI004 germline (control) and HCI004 PDX, and the average mapping quality of aligned reads from the sample (MQ, if no reads align MQ = 0), coverage standard deviation from 500 controls (Coverage SD, indicating common CNV), overlapping segmental duplications published by Bailey JA et al. 2002 (SEG-DUP, used as control for germline CNVs), discordant pairs (DP), split reads (SR), variants from the Database of Genomic Variants (DGV), and RefSeq genes (Genes)
Fig. 5
Mutational signature analysis of the PDXs. A Output mutational profiles of the six PDX models from deconstructSigs displaying the fraction of mutations found in each trinucleotide context B pie charts of the mutational signatures identified for each of the six PDX models from deconstructSigs
Using a stringent cutoff [18], we have created a list of high confidence copy number alterations and structural variants for each of the PDXs (Additional file 6: Table S11 and Additional file 7: Table S12). Recurrently amplified regions across the PDX models are chromosome 8q in HCI002/005/010/012 (covering MYC, Additional file 8: Table S13) and chromosome 7p in HCI004/HCI008 (covering EGFR). Frequent deleted regions harbour classic tumour suppressor genes, for example PTEN in HCI004/HCI010 (Fig. 4C, Additional file 6: Table S11). There are a few SV regions that harbouring breast cancer associated tumour suppressor genes, such as PTPRD in HCI008 model and CSMD3 in HCI012 model (Additional file 7: Table S12).
Because PDX tumours probably model more aggressive breast cancers (DeRose et al.), we proposed that genomic analysis of PDX models may reveal genes associated with very aggressive disease. Interestingly, the most frequently deleted gene in PDX models, CSMD1(in 3 out of our six PDX models), is much more frequently deleted in metastatic breast cancer (14%) than in early disease (2%) (Fig. 6A). Low expression of CSMD1 is also associated with poor survival outcome in the METABRIC early cancer cohort (Fig. 6B) and is especially associated with poor survival outcome in the LumB subtype (Additional file 1: Fig. S9). Interestingly, we identified copy number loss of CSMD1 in 23 of 32 PDXs in another PDX sequencing dataset [6]. These data suggest that CSMD1 plays a critical role in suppressing growth or survival of metastatic breast cancers.
Fig. 6
Genes with enriched genomic alterations in PDXs. A, B Genes with frequent copy number alterations in PDXs samples showing higher copy number variations in a metastatic breast cancer cohort than a primary breast cancer cohort (cbioportal). Each row indicates a gene and each column indicates a breast cancer sample. Copy number gains are in red and deletions are in blue. CSMD1, for example, showing a much high frequency of copy number deletion in the metastatic cohort. C Kaplan–Meier survival analysis of METABRIC discovery cohort samples stratified by CSMD1 expression status. Top 25% samples with high CSMD1 is in red, showing a better survival outcome compared to those with low CSMD1 expression
留言 (0)