

記住我






The corpus callosum (CC) is the greatest forebrain commissure in humans, containing approximately two hundred million callosal axons, and its major role is to integrate and transfer information between the two hemispheres. This interhemispheric communication is important for the functions of the cerebral cortex such as integration of sensory, motor, and visuomotor information, abstract reasoning, language processing and management of social and emotional stimuli (Paul et al., 2007; Edwards et al., 2014; Palmer and Mowat 2014; De Leon Reyes et al., 2020). The adult CC is anatomically divided into several regions: rostrum, genu, body, isthmus, and splenium. The rostrum and genu link the frontal and premotor regions of the cerebral cortex, the middle part connects the motor, somatosensory, and parietal regions, while the splenium conjoins the temporal and occipital cortices on both sides (De Leon Reyes et al., 2020).
The aim of this paper is to present the genetic abnormalities involved in the etiology of ACC and their relationship to CC development. We also focused on the suggestive associated features and the benefits of genetic testing for valuable prognostic information and the development of personalized management.
Literature search strategies and data collectionThe data synthesized and presented in this review was obtained by examining the literature (OMIM, PubMed, MEDLINE, Google Scholar databases) and using the following keywords: corpus callosum agenesis, corpus callosal disorders, corpus callosum development, genetic heterogeneity, genetic abnormalities, chromosomal abnormalities, monogenic disorders, chromosomal microarray analysis (CMA), whole exome sequencing (WES), whole genome sequencing (WGS), next generation sequencing (NGS). Only English language papers published within the previous 10 years were considered for this review. A narrative synthesis was generated for the main outcome. The Online Mendelian Inheritance in Man (OMIM) database was used as the reference for gene and mutation symbols.
Development of the corpus callosumCC development begins around the 12th week of gestation, and follows well-orchestrated events: neuronal and glial proliferation, neuronal migration and specification, midline patterning, axonal growth and guidance, and post-guidance refinement. The CC is complete and easily recognized by the 14–15th week of gestation whereas the splenium becomes prominent by the 18–19th week of gestation. CC development is a complex process and involves many pathways such as Semaphorin/Plexin/Neuropilin, Slit/Robo, Eph/Ephrin, Netrin/DCC/Unc5, Wnt/Ryk and FGF8/MAPK (De Leon Reyes et al., 2020; Ku and Torii 2020). Some molecules act as nodes of a transcriptional network regulating CC formation. Special AT-rich sequence-binding protein (SATB2), a nuclear matrix DNA-binding protein, involved in transcriptional regulation and chromatin remodeling, plays a central role in development of callosal projections. SATB2 participates in the direct transcriptional repression of CTIP2 gene (chicken ovalbumin upstream promoter transcription factor-interacting protein 2) (OMIM 606558), and promotes FEZF2 gene (Fez family zinc finger protein) (OMIM 607414) and SOX5 gene (OMIM 604975) expression in subcerebral neurons. FEZF2 in turn inhibits high-level SATB2 gene (OMIM 608148) expression (McKenna et al., 2015; Huang et al., 2020; Ku and Torii 2020). Expressions of SATB2 and CTIP2 genes are influenced by histone methylation (e. g. DOT1L), calcium signaling, and other mechanisms (Franz et al., 2019). Molecules from Netrin-1 signaling pathway also interact with SATB2 gene: DCC gene (OMIM 120470) is directly downregulated by SATB2, while UNC5C gene (OMIM 603610) is directly upregulated and downregulated by SATB2 and CTIP2 (Srinivasan et al., 2012). The heat shock cognate protein HSC70 is required for the stability of the DCC/TRIO signaling complex and to mediate axon guidance (DeGeer et al., 2015). Netrin-1 can trigger either attraction or repulsion of the axon depending on the receptor it binds. SATB2 also directly upregulates the expression of EPHA4 gene (OMIM 602118), a member of Eph/ephrin pathways (Srinivasan et al., 2012). Transcription factors FOXG1 promotes SATB2 gene expression and directly represses ROBO1 gene (roundabout guidance receptor 1) (OMIM 602430) and SLIT3 gene (OMIM 603745) expression (Cargnin et al., 2018). Slit molecules bind Robo receptors and act as a repulsive axon guidance cue (Gonda et al., 2020). Amyloid precursor protein (APP) serves as a Slit receptor and mediates axon repulsion (Wang et al., 2017). Semaphorins (SEMA3A and SEMA3C), also have essential roles in corpus callosum development, through signaling mediated by their coreceptors Neuropilin-1 and Plexin-A1 (Hossain et al., 2019).
Agenesis of the corpus callosum - epidemiology and causesMany factors could disrupt this complex developmental process and lead to agenesis of the corpus callosum (ACC) also known as corpus callosal disorders. ACC, complete or partial, is one of the most common cerebral anomaly in humans with a reported incidence of 1.8 per 10,000 livebirths, but a significant percentage of prenatally diagnosed cases result in termination of pregnancy or spontaneous abortion (Glass et al., 2008; Weise et al., 2019). The prevalence is higher in children with developmental disabilities: 230–600 per 10,000 (Schell-Apacik et al., 2008; Palmer and Mowat 2014). An increased risk for male gender and preterm birth has been reported (Ballardini et al., 2018). ACC may occur as an isolated anomaly or as a component of a complex disorder, caused by genetic abnormalities, teratogenic exposures or vascular factors. The prevalence of associated anomalies at birth varies considerably among studies, between 39.6% and 86.5% (Glass et al., 2008; Szabo et al., 2011; Stoll et al., 2019). According to Morris et al., 52% of ACC occur as an isolated condition, 25% have associated anomalies (unknown etiology), and 23% are syndromic (with chromosomal, monogenic or teratogenic causes) (Morris et al., 2019). The most common associated anomalies involve the musculoskeletal, the central nervous, the cardiovascular, the urogenital, and the digestive systems (Stoll et al., 2019). The central nervous system anomalies include hydrocephalus, cerebellar hypoplasia, periventricular nodular heterotopia, polymicrogyria, microgyria, and lissencephaly (Glass et al., 2008; Ballardini et al., 2018; Stoll et al., 2019). In cases with hydrocephalus, the differentiation between arrested ventriculomegaly and progressive hydrocephalus is essential for appropriate management. Based on neurological, ophthalmological, and psychometric evaluations, this distinction is very difficult. Radiological markers (frontal-occipital horn ratio, apparent diffusion coefficient, and cerebral blood flow) and intracranial pressure monitoring are very useful diagnostic tools (Leliefeld et al., 2010; Hurni et al., 2018). Genetic causes include complete or partial chromosomal anomalies, autosomal dominant, autosomal recessive or X-linked monogenic disorders, de novo or inherited. Several selected syndromes are discussed in more detail concerning their associated anomalies. Multiple other genetic abnormalities are summarized in Tables 1 and 2.
TABLE 1
TABLE 2
Chromosomal anomaliesChromosomal abnormalities have been identified in 4.81–17.8% of ACC; especially when associated congenital anomalies are present. The reported complete aneuploidies include: trisomy 18, 13, 8 in mosaic, 21 and X monosomy (Glass et al., 2008; O'Driscoll et al., 2010; Palmer and Mowat 2014; D'Antonio et al., 2016). Callosal abnormalities are also associated with partial chromosomal anomalies, the most common: 8p inverted duplication/deletion syndrome, 1q43q44, 6q27 and 17q21.31 microdeletions are characterized bellow. However, many copy number variants have been reported in cases with ACC (Table 1).
1q43q44 microdeletion syndrome1q43q44 microdeletion syndrome (OMIM 621337) is a disorder characterized by moderate to severe intellectual disability with limited or no expressive speech, microcephaly, agenesis/hypogenesis of the CC, seizures, suggestive facial dysmorphism (round face, prominent forehead, hypertelorism, epicanthal folds, flat nasal bridge, and malformed and low-set ears), and hand and foot anomalies. Balliff et al. proposed a 75 Kb critical region for ACC that contains ZBTB18 gene (OMIM 608433), which encodes a transcriptional repressor of key genes involved in neuronal development (Ballif et al., 2012). The phenotypic heterogeneity (no ACC) in cases with critical region deletion has been explained by incomplete penetrance or variable expressivity. Depienne et al. considered that ACC is also influenced by the alteration of neighboring genes especially HNRNPU (OMIM 617391) (Ballif et al., 2012; de Munnik et al., 2014; Cohen et al., 2017; Depienne et al., 2017).
8p inverted duplication/deletion syndrome [invdupdel (8p)]Interstitial inverted duplication 8p associated with 8pter deletion is a complex and rare chromosomal rearrangement. Clinical features include mild to severe intellectual disability, hypoplasia/agenesis of the CC, characteristic facial dysmorphism (prominent forehead, temporal baldness, anteverted nostrils, eversion of the lower lip, large mouth and ears), skeletal anomalies (scoliosis/kyphosis), hypotonia, and congenital heart defects. The invdupdel (8p) consists of a deletion of the telomeric region (8p23-pter) followed by an intermediate intact fragment, and a proximal inverted duplication of variable size. Rearrangements are mediated by two highly repetitive regions: the olfactory receptor gene clusters or defensin repeat (ORDRs); the polymorphic 8p23 inversion between these clusters increases the susceptibility of 8p to rearrangements. The 8p23.2-pter region contains the genes ARHGEF10 (OMIM 608236) and CSMD1 (OMIM 608397), associated to central nervous system development (de Die-Smulders et al., 1995; Giglio et al., 2001; Garcia-Santiago et al., 2015; Redaelli et al., 2022).
6q27 microdeletion6q27 deletion is associated with a variable phenotype including intellectual disability, seizure, hypotonia, brain malformations (ACC, periventricular nodular heterotopia (PNH), polymicrogyria, hydrocephalus, and cerebellar malformations), short neck and craniofacial dysmorphism (microcephaly, broad nose with prominent nasal root and bulbous nasal tip, large and low-set ears, and downturned mouth). Other commonly reported findings are: joint laxity, heart defects and retinal anomalies. The size of deletion varies between 0.4 Mb and 10.8 Mb and does not seem to correlate with phenotype severity, suggesting the existence of a minimal critical region. Hanna et al. proposed a 325 kb critical region for brain malformation and intellectual disability, comprising the DLL1 (OMIM 606582), PSMB1 (OMIM 602017), TBP (OMIM 600075), and PDCD2 (OMIM 600866) genes (Hanna et al., 2019). The key role of DLL1 is supported by cases with brain anomalies, intellectual disability and pathogenic variants in DLL1 gene. DLL1 gene is Notch ligand with an important role in development of the central nervous system (regulates neuronal differentiation), somites and lymphocytes (Striano et al., 2006; Peddibhotla et al., 2015; Fischer-Zirnsak et al., 2019; Hanna et al., 2019; Lesieur-Sebellin et al., 2022).
17q21.31 microdeletion17q21.31 microdeletion known as Koolen-de Vries syndrome (KDVS) (OMIM 610443) is characterized by intellectual disability, hypotonia, suggestive facial dysmorphism (upslanting palpebral fissures, blepharophimosis, ptosis, epicanthus, a pear-shaped nose with bulbous nasal tip, and large/protruding ears), friendly behavior, brain anomalies (ventriculomegaly, ACC, hydrocephalus, Arnold-Chiari type I malformation), seizures, joint hypermobility, heart defects, and genitourinary anomalies. KDVS is caused by a 17q21.31 microdeletion or by a pathogenic variant in the KANSL1 gene (OMIM 612452). Genotype-phenotype correlation studies did not report significant clinical differences between microdeletions and pathogenic variant patients. The recurrent 17q21.31 microdeletion breakpoints map to flanking extensive low copy repeats suggesting that deletion is mediated by non-allelic homologous recombination. KANSL1 encodes the KAT8 regulatory NSL complex Subunit 1, a member of a histone acetyltransferase complex, which regulates global transcription (Koolen et al., 2008; Zollino et al., 2012; Koolen et al., 2016).
Monogenic disordersMonogenic causes have been identified in 8–35% of ACC (Bedeschi et al., 2006; Schell-Apacik et al., 2008; Palmer and Mowat 2014; Ballardini et al., 2018; Morris et al., 2019). Advances in genetic sequencing technologies have enabled the discovery of pathogenic variants in a large number of genes: from those involved in development of callosal projections to genes encoding components of microtubules or metabolic enzymes (Table 2) (Schell-Apacik et al., 2008; Palmer and Mowat 2014; Ballardini et al., 2018; Morris et al., 2019).
Acrocallosal syndrome (ACS)ACS (OMIM 200990) is a ciliopathy characterized by ACC, distal limb anomalies (postaxial polydactyly of the hands, and preaxial polydactyly of the feet), intellectual disability and facial dysmorphism (prominent forehead, hypertelorism, short nose with broad nasal bridge). In ACS the most frequent mutations were found in KIF7 gene (15q26.1) (OMIM 611254) while mutations of GLI3 gene (7p14.1) (OMIM 165240) are rare (Elson et al., 2002; Putoux et al., 2011). Both genes are involved in the ciliary sonic hedgehog (SHH) pathway. Depletion of proteins related to SHH pathway is associated with ciliogenesis defects. KIF7 gene encodes a member of kinesin motor proteins family, that localize from the base to the tip of the cilium and plays important roles in the regulation of microtubule acetylation and stabilization, and SHH signaling (Niceta et al., 2020). GLI3 gene encodes a zinc finger transcription factor, that interacts with microtubule associated MID1 protein and KIF7 and has a dual function: as a transcriptional activator and a repressor of SHH pathway (Laclef et al., 2015). Biallelic mutations in KIF7 gene have been reported in Hydrolethalus syndrome and rare cases of Joubert syndrome, with overlapping features, suggesting a continuous phenotypic spectrum. A severe phenotype is associated with KIF 7 gene truncating mutations, while missense KIF7 gene variants have been reported in milder cases (Putoux et al., 2011). Asadollahi et al. reported mutation in KIF7, SHH, and CPLANE1 genes in patients with suggestive facial dysmorphism for ACS and ciliopathy features (Asadollahi et al., 2018). Monoallelic mutations in GLI3 gene have been reported in Greig cephalopolysyndactyly and Pallister Hall syndromes. A group of ciliopathies associate intellectual disability and brain malformations, including ACC, highlighting the critical role for primary cilia in brain development (Valente et al., 2014; Arghir et al., 2021; Hasenpusch-Theil and Theil 2021).
TubulinopathiesTubulinopathies comprise a wide spectrum of brain malformations (lissencephaly, cerebellar hypoplasia, ACC, pachygyria, dysgyria), variable degree of motor and intellectual disabilities and epilepsy, caused by pathogenic variants in tubulin genes (α, β and γ isotypes) (Romero et al., 2018). The alpha and beta families of tubulin genes encode proteins which form heterodimers as fundamental components of microtubules. Tubulins interact with other proteins (e.g. β-III-tubulin connects with DCC in a netrin-1 dependent manner) and play key roles in neuronogenesis, neuronal migration, cortical organization–pial basement membrane integrity, and also in axon guidance (Qu et al., 2013; Boyer and Gupton 2018). All mutations reported in TUBA1A (12q13.12, OMIM 602529), TUBB2B (6p25.2, OMIM 612850), TUBB3 (16q24.3, 602,661) and TUBB (6p21.33, 1919130) genes are heterozygote missense variants; this could suggest a dominant-negative effect. TUBA1A gene missense variants are the most frequent; some hot spots have been reported: Arg264 (c.790C>T, predominantly results in central pachygyria, whereas c.791G>A causes microlissencephaly with complete agenesis of the corpus callosum), Arg402 and Arg422 (Romaniello et al., 2015; Hebebrand et al., 2019).
FOXG1 syndromeFOXG1 syndrome (OMIM 613454), is a rare neurodevelopmental disorder, with a wide spectrum of features ranging from severe intellectual disability with poor or absent speech development, postnatal microcephaly, corpus callosum anomalies (agenesis/hypogenesis), delayed myelination, seizures, and dyskinesia, to deficient social interactions, disrupted circadian rhythm, and autistic traits. It has been previously classified as a congenital variant of Rett syndrome, due to clinical features and evolution, including a normal perinatal period followed by a phase of developmental regression at the age of 3–6 months. FOXG1 gene (OMIM 164874), located at 14q12, encodes the forkhead box protein G1, a conserved transcriptional repressor, that play a critical role in forebrain development, from the timing of neurogenesis, to the patterning of the cerebral cortex and callosal projections. FOXG1 gene truncating mutations (frameshift or nonsense) in the N-terminal domain and 14q12 microdeletions associate a severe phenotype, while FOXG1 gene missense mutations in the forkhead domain lead to milder phenotype (Kortum et al., 2011; Leombroni et al., 2018; Mitter et al., 2018; Hou et al., 2020; Akol et al., 2022).
ACC–Neuropathy syndrome (Andermann syndrome)Hereditary motor and sensory neuropathy with ACC (OMIM 218000) is a channelopathy characterized by severe progressive sensorimotor neuropathy with resulting hypotonia, areflexia, and amyotrophy, and by dysgenesis of the corpus callosum. Common features include intellectual and developmental delays, psychiatric manifestations (paranoid elements, depression, hallucinations), progressive scoliosis and facial dysmorphism (palpebral ptosis, hypertelorism, hypoplastic maxilla, high-arched palate). It is inherited in an autosomal recessive manner, and the high incidence in the Saguenay and Lac-Saint-Jean regions of Quebec, Canada, due to the c.2436+1delG mutation in SLC12A6 gene (OMIM 604878) suggest a founder effect. SLC12A6 gene, located at 15q14, encodes a solute carrier family 12 member 6 (KCC3), a potassium chloride cotransporter, expressed in the peripheral and central nervous system. Impaired KCC3 function alters maintenance of homeostasis of the membrane threshold, leading to neuronal degeneration. A case with a gain of function mutation and peripheral neuropathy but no corpus callosum abnormalities has been reported (Gauvreau et al., 1993; Kahle et al., 2016; Flores et al., 2019).
Aicardi syndromeAicardi syndrome (OMIM 304050) is characterized by a triad of features: agenesis or hypogenesis of corpus callosum, chorioretinal lacunae, and infantile spasms. Other common findings include: periventricular and subcortical heterotopia, intracranial cysts, polymicrogyria, retinal and optic nerve colobomas, microphthalmia, costovertebral anomalies, gastrointestinal disorders (reflux, constipation, feeding difficulties) and suggestive facial dysmorphism (prominent premaxilla, upturned nasal tip, decreased angle of the nasal bridge, and sparse lateral eyebrows). It affects usually females, but the disease was identified in few cases with Klinefelter syndrome (47, XXY) or 46, XY males with profound microcephaly. The inheritance of Aicardi syndrome is considered X-linked dominant with early embryonic lethality in the majority of hemizygous males. Skewed X chromosome inactivation and translocations involving Xp22 have been reported in several cases but a candidate gene has not been identified. Nemos et al. excluded mutations in the CDKL5 gene (Xp22.13) (OMIM 300203) (Eble et al., 2009; Nemos et al., 2009; Wong and Sutton 2018).
Corpus callosum agenesis - Abnormal genitalia syndrome (Proud syndrome)Corpus callosum agenesis - abnormal genitalia syndrome (OMIM 300004) is characterized by ACC, neurological manifestations (intellectual disability, developmental delay, epilepsy, spasticity), and urogenital anomalies (hypospadias, cryptorchidism, renal dysplasia). It is part of a broad phenotypic spectrum of disorders caused by mutation in the ARX gene (OMIM 300382), ranging from lissencephaly to ACC with abnormal genitalia to developmental and epileptic encephalopathy without brain malformations to syndromic and nonsyndromic X-linked intellectual disability. ARX gene, located at Xp22.13, encodes a transcription factor which belongs to the class of homeobox proteins. ARX participate to fundamental processes of brain development: patterning, neuronal proliferation and migration, cell maturation and differentiation, axonal outgrowth and connectivity. ARX contains several domains, including the homeobox domain, a PRD-like domain, a N-terminal octapeptide domain, a central acidic domain, a C-terminal aristaless domain, as well as three nuclear localization sequences and four polyalanine tracts. Premature termination mutations and missense variants in the homeodomain or nuclear localization sequences are associated with severe phenotype involving brain malformations. Missense mutations outside of the homeobox domain or expansions in the polyalanine tracts lead to milder phenotype without brain malformation. Usually, males are severely affected, and heterozygous females may be unaffected or have a milder phenotype (Kato et al., 2004; Friocourt et al., 2006; Olivetti and Noebels 2012; Ghadami and Eshaghkhani 2019; Leung et al., 2022).
Pyruvate dehydrogenase complex (PDC) deficiencyPDC deficiency (OMIM 312170, 614111, 245348, 245349, 246904, 608782) is an inborn error of mitochondrial energy metabolism, with a wide phenotypic spectrum ranging from fatal neonatal lactic acidosis to neurological manifestations (hypotonia, seizures, ataxia and developmental delay) with structural brain abnormalities. ACC is found in over one-third of patients with PDHA1 gene mutations (Pavlu-Pereira et al., 2020; Savvidou et al., 2022). The disorder can result from mutations in multiple genes that encode components of the PDC: PDHA1 (Xp22.1) (OMIM 300502), PDHB (3p14.3) (OMIM 179060), DLAT (11q23.1) (OMIM 608770), PDHX (11p13) (OMIM 608769), DLD (7q31.1) (OMIM 238331) and PDP1 (8q22.1) (OMIM 605993). The most common are mutations in the PDHA1 gene, which encodes E1 alpha subunit, and has eleven exons. Frameshift PDHA1 gene mutations occur preferentially in exons 10 and 11, associate a very low enzyme activity and severe phenotype. Missense PDHA1 gene mutations are located usually in exons one to nine and are more prevalent in males. Hemizygous males are generally symptomatic and more severely affected (with significant lethality in infancy) whereas heterozygous females present variable clinical manifestations and greater survival due to random patterns of X-inactivation (Patel et al., 2012; Horga et al., 2019; Pavlu-Pereira et al., 2020).
Pyridoxine-dependent epilepsy (PDE)PDE (OMIM 266100) is characterized by recurrent seizures, usually with early onset, resistant to standard anticonvulsants but with clinical and electroencephalogram (EEG) improvement following pyridoxine supplementation. Intellectual disability is common (75%–80%) and does not correlate with seizure control or age at treatment initiation (Gospe et al., 2021; Coughlin et al., 2019). Callosal abnormalities are the most frequent neuroimaging findings and are not explained by treatment lag (Toldo et al., 2018; Tseng et al., 2022). PDE is caused by mutation in ALDH7A1 gene (OMIM 107323), located on 5q23.2, which encodes alpha-aminoadipic semialdehyde (α-AASA) dehydrogenase, a key enzyme in lysine metabolism. The enzyme deficiency, inherited in an autosomal recessive manner, leads to accumulation of α-AASA, pipecolic acid, and Δ-1-piperideine-6 carboxylate, responsible for sequestration of pyridoxal-5-phospate. Missense mutations are the most common and are more frequent in exons 5 and 6 (within the linker domain), 10 (within the NAD binding domain), 15 (within the catalytic domain). The most prevalent mutation is c.1279G>C (pGlu427Gln) and homozygotes patients present variable phenotype with neonatal or late-onset. Splice site (e.g. c.834G>A), frameshift and nonsense mutations have been also reported (Coughlin et al., 2019). The deep intronic mutation c.696–502G>C results in the usage of two cryptic splice sites and introduction of a pseudoexon between exons 7 and 8 (Milh et al., 2012). Coughlin et al. hypothesized that some mutations affect not only the enzyme activity but also the stability or the ability to reach its targeted environment (Coughlin et al., 2019).
Congenital mirror movements (CMM)CMM (OMIM 157600) is characterized by early onset, involuntary movements of one side of the body that mirror intentional movements on the opposite side. Physiologic mild mirror movements are occasionally found in young children, but their persistence beyond the age of 7 years is pathologic. CMM usually persist throughout life and involve predominantly the upper limbs, more severe distal (Meneret et al., 2014). Monoallelic truncating (frameshift and nonsense) mutations of DCC gene (OMIM 120470) are associated with CMM. Missense mutations c.2378T>G (p.Val793Gly) and c.2414G>A (p.Gly805Glu), located within the NTN1 binding interface are also associated with CMM. Monoallelic DCC gene mutations are associated with variable expressivity and incomplete penetrance. The DCC gene, located at 18q21.2, encodes a functional receptor for netrin (NTN1), a transmembrane glycoprotein belonging to the immunoglobulin (Ig) superfamily of cell adhesion molecules. DCC plays a key role in the transduction of NTN1-induced attractive and repulsive signaling in the coordinated outgrowth and guidance of commissural axons that cross the anatomical midline of the body. Corticospinal axons and callosal axons use slightly different signaling pathways, and a DCC mutation may differentially affect commissural versus subcerebral axon trajectories, explaining the variable phenotypes: mirror movements, ACC, or both (Srour et al., 2010; Marsh et al., 2018). CMM is also caused by monoallelic mutation in NTN1 gene (17p13.1) (OMIM 601614), RAD51 gene (15q15.1) (OMIM 179617) and biallelic mutations in DNAL4 gene (22q13.1) (OMIM 610565), but no callosal anomalies have yet been reported (Ahmed et al., 2014; Meneret et al., 2014).
Horizontal gaze palsy with progressive scoliosis (Split brain syndrome) (OMIM 617542)Biallelic, DCC gene mutations lead to a complex syndrome associated with a broad disorganization of white-matter tracts throughout the CNS (Jamuar et al., 2017). The features include absence of all commissures (including the CC, anterior and posterior), brainstem defects (including hypoplasia of the pons and midbrain), horizontal gaze palsy, impaired intellectual development and progressive scoliosis. Biallelic mutations are associated with complete penetrance (Jamuar et al., 2017; Marsh et al., 2018). Biallelic mutation in ROBO3 gene (11q24.2) (OMIM 608630), which encodes a coreceptor of NTN1, leads to horizontal gaze palsy with progressive scoliosis and brainstem defects (hypoplasia of the pons and cerebellar peduncles, butterfly configuration of the medulla, and pontine cleft in the midline); no CC anomalies have been reported. ROBO3 is a member of the roundabout family of receptors, inhibits ROBO1/2, interacts with DCC and potentiate commissural axonal outgrowth and attraction (Jen et al., 2004; Pinero-Pinto et al., 2020).
Septo-optic dysplasia (SOD)SOD (OMIM 182230) is characterized by a clinical triad of optic nerve hypoplasia, dysfunction of the hypothalamic-pituitary axis, and midline brain abnormalities, including agenesis of the septum pellucidum and CC. For a SOD spectrum diagnosis, a combination of any two of the three features is required. Only 30%–47% of patients present the complete classical triad. Additional findings include developmental delay, seizures, cerebral palsy, autism, sleep disorders, thermoregulatory disturbances, hearing loss, anosmia, and heart defects. Associated brain abnormalities (hydrocephalus, polymicrogyria, grey matter heterotopias) have also been reported (sometimes referred to as SOD-plus syndrome) (Webb and Dattani 2010; Ganau et al., 2019; Ward et al., 2021). SOD is usually sporadic but familial cases with intrafamilial variability have been reported. Genetic abnormalities have been identified in <1% of the patients. Homozygous or heterozygous mutations in HESX1 (3p14.3) (OMIM 601802) have been described in patients with variable phenotypes ranging from isolated growth hormone deficiency to SOD. HESX1 encodes a paired-like homeobox transcription factor, which acts as a transcriptional repressor and is essential for normal forebrain and pituitary development. Heterozygous HESX1 mutations are associated with a milder phenotype and incomplete penetrance (Kelberman and Dattani 2008; Webb and Dattani 2010; Fang et al., 2016). Mutations in other genes involved in the regulation of the Wnt/β-catenin signaling pathway have been reported in SOD cases: SOX2 (3q26.33) (OMIM 184429), SOX3 (Xq27.1) (OMIM 313430), OTX2 (14q22.3) (OMIM 600037), TCF7L1 (2p11.2) (OMIM 604652) or TAX1BP3 (17p13.2) (OMIM 616484). SOX2 mutations are associated with severe eye abnormalities, and additional features (genital anomalies, esophageal atresia, intellectual disability, seizures) (Blackburn et al., 2018). SOX3 duplications/point mutations have been reported in cases with hypopituitarism and/or intellectual disability (Arya et al., 2019; Li et al., 2022). OTX2 pathogenic variants are associated with hypopituitarism and eye abnormalities and incomplete penetrance (Gregory et al., 2021). Mutations in genes involved in hypogonadotropic hypogonadism with or without anosmia (OMIM 612702) - FGFR1 (8p11.23) (OMIM136350) and FGF8 (10q24.32) (OMIM 600483) have also been described in SOD patients suggesting a genetic overlap (Raivio et al., 2012). The etiology of SOD remains unclear in the majority of patients; vascular abnormalities, teratogens, and young maternal age may also be involved (Ganau et al., 2019).
Isolated ACCThe genetic causes of isolated ACC appear more elusive but some pathogenic variants in genes associated with syndromic ACC have been reported. DCC gene missense mutations, located within the NTN1 binding sites (in fourth, fifth and sixth fibronectin type III-like domains), are associated with isolated ACC, in females. Marsh et al. observed a significant dose-dependent increase in DCC gene expression in testosterone-treated neural stem cells derived from human embryonic stem cells and suggested that isolated ACC may occur when DCC gene expression is below a critical level during CC development. Jouan et al. identified compound heterozygous missense variants in the CDK5RAP2 gene (OMIM 608201) in 3 sibs with isolated ACC. The CDK5RAP2 gene, located at 9q33.2, encodes cyclin-dependent kinase 5 regulatory subunit 2 (CDK5RAP2) a pericentriolar structural protein that has a major role in the microtubule-organizing function of the centrosome through interaction with the γ-tubulin ring complex. Its expression is particularly high within the brain, especially within the thalamus and corpus callosum (Jouan et al., 2016). These findings suggested that alteration of CDK5RAP2 activity may dysregulate neurogenic cell divisions. Biallelic mutations in CDK5RAP2 are associated with autosomal recessive primary microcephaly and isolated ACC. Jouan et al. hypothesized that mutations with residual function may lead to isolated ACC, while severe loss-of-function variants (nonsense, splice site, truncating missense) may result in severe microcephaly with reduced brain size and concomitantly with partial or complete ACC (Jouan et al., 2016). The factors determining the phenotypic variability are complex and include the type and location of mutation and possible genetic modifiers (Jouan et al., 2016; Marsh et al., 2018).
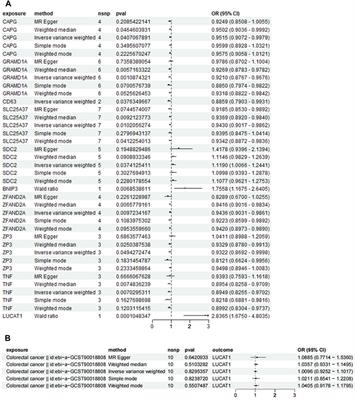
The large number of partial chromosomal anomalies and monogenic syndromes associated with ACC illustrate the high genetic heterogeneity (Figure 1).
FIGURE 1
FIGURE 1. Chromosome ideograms illustrating genetic heterogeneity: partial chromosomal anomalies are represented on the left-hand side of the ideogram; genes associated with ACC are represented on the right-hand side of the ideogram (created in BioRender–publication license May-30–2022); (Chromosome 1: ARID1A, GPSM2, KIF14, FH. Chromosome 2: SOX11, NPHP1, ZEB2, LRP2. Chromosome 3: HESX1, PDHB, ZBTB20, NPHP3. Chromosome 4: CC2D2A. Chromosome 5: CPLANE1, ALDH7A1, TXNDC15, NSD1. Chromosome 6: TUBB2B, SOX4, TUBB, AHI1, ARID1B. Chromosome 7: ACTB, GLI3, DLD. Chromosome 8: CSPP1, TMEM67, PDP1. Chromosome 9: CDK5RAP2, INPP5E. Chromosome 10: FGFR2. Chromosome 11: PDHX, DPF2, TMEM216, DHCR7, DLAT, HYLS1, ROB O 3. Chromosome 12: C12orf57, ARID2, TUBA1A, SMARCC2, CEP290, TCTN2. Chromosome 14: FOXG1, KIAA0586. Chromosome 15: SLC12A6, RAD51, KIF7. Chromosome 16: CREBBP, RPGRIP1L, TMEM231, TUBB3. Chromosome 17: TMEM107, NTN1, B9D1, SMARCE1, MKS1, ACTG1. Chromosome 18: EPG5, DCC, PIGN, RTTN. Chromosome 19: SMARCA4, B9D2. Chromosome 22: SMARCB1, DNAL4, EP300. Chromosome X: CDKL5, ARX, PDHA1, EFNB1, MED12, L1CAM).
PrognosisIn isolated ACC, neurodevelopmental outcomes range from normal development in about 64.8–75% of the individuals to different levels of intellectual disability, including severe (in 12% of cases) (D'Antonio et al., 2016; Bernardes da Cunha et al., 2021). Sleep disturbances are also common and include: greater sleep onset delay, decreased sleep duration, sleep anxiety, night wakings, parasomnias, sleep-disordered breathing, daytime sleepiness, increased slow-wave sleep, more distressful dreams, and narcolepsy (Ingram and Churchill 2017; Yates et al., 2018). Regular follow-up is necessary because some cases with initial “normal development” presented subtle speech, attention, and reasoning difficulties with increasing age. In a significant percentage of cases (approximately 40%) with ACC, social and behavioral problems have been reported: emotional immaturity, impaired social competence, deficits in planning, and poor communication of emotions (Wegiel et al., 2017). While autistic elements have been reported in cases with ACC, MRI in patients with autism revealed thinning of corpus callosum but further studies are necessary to elucidate the connection between corpus callosum anomalies and autism (Frazier and Hardan 2009; Paul et al., 2014; Hofman et al., 2020). Other adverse outcomes related to ACC include epilepsy, visual problems and psychiatric disorders (schizophrenia) (Popoola et al., 2019). The rate of severe disability in cases with associated anomalies is higher than that in patients with isolated ACC. The spectrum of neurodevelopmental outcome depends on several factors, such as the extension of the ACC (complete or partial), the presence and severity of associated defects, as well as on the magnitude of residual interhemispheric transfer through other commissures (Guadarrama-Ortiz et al., 2020). A checklist of suggested diagnostic evaluations in a postnatal patient with ACC is summarized in Table 3.
TABLE 3
TABLE 3. Suggested diagnostic evaluations in a postnatal patient with ACC.
The variable neurodevelopmental outcomes of ACC make prenatal counseling a challenge. Fetal MRI plays a critical role in detection of associated anomalies, a major prognosis determinant (Bernardes da Cunha et al., 2021). Some studies reported discrepancy between prenatal diagnosis of isolated ACC and postnatal or postmortem diagnosis of additional anomalies (5% in complete ACC and 15% in partial ACC). These discrepancies may be related to the technology, method of diagnosis, or to the timing of investigations because some anomalies may be identified only in the third trimester of pregnancy (Paladini et al., 2013; Griffiths et al., 2017; Leombroni et al., 2018).
Prenatal genetic diagnosis is essential in these cases. Chromosomal abnormalities (except mosaicism if abnormal cells are not present in the examined specimen) and an increasing number of monogenic disorders can be diagnosed prenatally. Conventional prenatal karyotype identifies complete or large partial aneuploidies in 4.8%–7.5% of ACC (Leombroni et al., 2018). Prenatal chromosomal microarray analysis (CMA) in cases with normal karyotype reveals pathological copy number variations in 5.7–6.9% of ACC (Greenbaum et al., 2021). However, the diagnostic rate of fetal karyotype and CMA does not exceed 15–20%, because monogenic causes have the highest frequency. In this context, many studies highlighted the utility of WES in determining the genetic etiology of CCA including in prenatal cases. The detection rate is higher in cases with associated anomalies. WES can also detect many copy number variations. Gene variant interpretation could be challenging due to incomplete prenatal phenotype (Reches et al., 2018; Heide et al., 2020; Lei et al., 2021; She et al., 2021).
ConclusionIntensive research over the last decade has highlighted the genetic heterogeneity of ACC, which reflects the complexity of CC development. Although an impressive number of chromosomal abnormalities and gene mutations have been reported in ACC–associated syndromes, there are still conditions of unclear etiology. Incomplete penetrance and variable expressivity, unexplained by the type of mutation, suggest the existence of genetic modifiers. Detection of associated anomalies and genetic causes is essential in prenatal detected cases, given the variable outcome, oscillating between the normal cognitive level and severe psychomotor delay. WES alone or simultaneous, WES and CMA may be considered a first-tier test in the prenatal diagnosis of ACC given the frequency of monogenic and chromosomal anomalies and the time constraints. The elucidation of all molecular mechanisms of ACC and the genotype-phenotype correlations will aid in understanding of CC role and in development of personalized management.
Author contributionsAll authors contributed equally to this paper. MP, SP, AL wrote the initial manuscript, CG, VL, VG reviewed and improved the article.
Conflict of interestThe authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s noteAll claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ReferencesAhmed, I., Mittal, K., Sheikh, T. I., Vasli, N., Rafiq, M. A., Mikhailov, A., et al. (2014). Identification of a homozygous splice site mutation in the dynein axonemal light chain 4 gene on 22q13.1 in a large consanguineous family from Pakistan with congenital mirror movement disorder. Hum. Genet. 133 (11), 1419–1429. doi:10.1007/s00439-014-1475-8
PubMed Abstract | CrossRef Full Text | Google Scholar
Akol, I., Vogel, F. T., and Vogel, T. (2022). Paving therapeutic avenues for FOXG1 syndrome: Untangling genotypes and phenotypes from a molecular perspective. Ijms 23 (2), 954. doi:10.3390/ijms23020954
CrossRef Full Text | Google Scholar
Arghir, A., Popescu, R., Resmerita, I., Budisteanu, M., Butnariu, L. I., Gorduza, E. V., et al. (2021). Pallister-killian syndrome versus trisomy 12p-A clinical study of 5 new cases and a literature review. Genes 12 (6), 811. doi:10.3390/genes12060811
PubMed Abstract | CrossRef Full Text | Google Scholar
Arya, V. B., Chawla, G., Nambisan, A. K. R., Muhi-Iddin, N., Vamvakiti, E., Ajzensztejn, M., et al. (2019). Xq27.1 duplication encompassing SOX3: Variable phenotype and smallest duplication associated with hypopituitarism to date - a large case series of unrelated patients and a literature review. Horm. Res. Paediatr. 92 (6), 382–389. doi:10.1159/000503784
PubMed Abstract | CrossRef Full Text | Google Scholar
Asadollahi, R., Strauss, J. E., Zenker, M., Beuing, O., Edvardson, S., Elpeleg, O., et al. (2018). Clinical and experimental evidence suggest a link between KIF7 and C5orf42-related ciliopathies through Sonic Hedgehog signaling. Eur. J. Hum. Genet. 26 (2), 197–209. doi:10.1038/s41431-017-0019-9
PubMed Abstract | CrossRef Full Text | Google Scholar
Ballardini, E., Marino, P., Maietti, E., Neville, G. A. J., and Neville, A. J. (2018). Prevalence and associated factors for agenesis of corpus callosum in Emilia Romagna (1981-2015). Eur. J. Med. Genet. 61 (9), 524–530. doi:10.1016/j.ejmg.2018.06.004
PubMed Abstract | CrossRef Full Text | Google Scholar
Ballif, B. C., Rosenfeld, J. A., Traylor, R., Theisen, A., Bader, P. I., Ladda, R. L., et al. (2012). High-resolution array CGH defines critical regions and candidate genes for microcephaly, abnormalities of the corpus callosum, and seizure phenotypes in patients with microdeletions of 1q43q44. Hum. Genet. 131 (1), 145–156. doi:10.1007/s00439-011-1073-y
PubMed Abstract | CrossRef Full Text | Google Scholar
Bedeschi, M. F., Bonaglia, M. C., Grasso, R., Pellegri, A., Garghentino, R. R., Battaglia, M. A., et al. (2006). Agenesis of the corpus callosum: Clinical and genetic study in 63 young patients. Pediatr. Neurol. 34 (3), 186–193. doi:10.1016/j.pediatrneurol.2005.08.008
PubMed Abstract | CrossRef Full Text | Google Scholar
Bernardes da Cunha, S., Carneiro, M. C., Miguel Sa, M., Pina, A. C., and Pina, C. (2021). Neurodevelopmental outcomes following prenatal diagnosis of isolated corpus callosum agenesis: A systematic review. Fetal Diagn Ther. 48 (2), 88–95. doi:10.1159/000512534
PubMed Abstract | CrossRef Full Text | Google Scholar
Blackburn, P. R., Chacon-Camacho, O. F., Ortiz-González, X. R., Reyes, M., Lopez-Uriarte, G. A., Zarei, S., et al. (2018). Extension of the mutational and clinical spectrum of SOX2 related disorders: Description of six new cases and a novel association with suprasellar teratoma. Am. J. Med. Genet. 176 (12), 2710–2719. doi:10.1002/ajmg.a.40644
PubMed Abstract | CrossRef Full Text | Google Scholar
Cargnin, F., Kwon, J. S., Katzman, S., Chen, B., Lee, J. W. S.-K., and Lee, S. K. (2018). FOXG1 orchestrates neocortical organization and cortico-cortical connections. Neuron 100 (5), 1083–1096. doi:10.1016/j.neuron.2018.10.016
PubMed Abstract | CrossRef Full Text | Google Scholar
Cohen, J. S., Srivastava, S., Farwell Hagman, K. D., Shinde, D. N., Huether, R., Darcy, D., et al. (2017). Further evidence thatde novomissense and truncating variants inZBTB18cause intellectual disability with variable features. Clin. Genet. 91 (5), 697–707. doi:10.1111/cge.12861
PubMed Abstract | CrossRef Full Text | Google Scholar
Coughlin, C. R., Spector, E., Meeks, N. J. L., Kronquist, K. E., Aslamy, M., Wempe, M. F., et al. (2019). The genotypic spectrum of ALDH7A1 mutations resulting in pyridoxine dependent epilepsy: A common epileptic encephalopathy. J. Inherit. Metab. Dis. 42 (2), 353–361. doi:10.1002/jimd.12045
PubMed Abstract | CrossRef Full Text | Google Scholar
D’Antonio, F., Pagani, G., Familiari, A., Khalil, A., Sagies, T. L., Malinger, G., et al. (2016). Outcomes associated with isolated agenesis of the corpus callosum: A meta-analysis. Pediatrics 138 (3), 445. doi:10.1542/peds.2016-0445
PubMed Abstract | CrossRef Full Text | Google Scholar
de Die-Smulders, C. E., Engelen, J. J., Schrander-Stumpel, C. T., Govaerts, L. C., de Vries, B., Vles, J. S., et al. (1995). Inversion duplication of the short arm of chromosome 8: Clinical data on seven patients and review of the literature. Am. J. Med. Genet. 59 (3), 369–374. doi:10.1002/ajmg.1320590318
PubMed Abstract | CrossRef Full Text | Google Scholar
de Munnik, S. A., García-Miñaúr, S., Hoischen, A., van Bon, B. W., Boycott, K. M., Schoots, J., et al. (2014). A de novo non-sense mutation in ZBTB18 in a patient with features of the 1q43q44 microdeletion syndrome. Eur. J. Hum. Genet. 22 (6), 844–846. doi:10.1038/ejhg.2013.249
PubMed Abstract | CrossRef Full Text | Google Scholar
DeGeer, J., Kaplan, A., Mattar, P., Morabito, M., Stochaj, U., Kennedy, T. E., et al. (2015). Hsc70 chaperone activity underlies Trio GEF function in axon growth and guidance induced by netrin-1. J. Cell Biol. 210 (5), 817–832. doi:10.1083/jcb.201505084
PubMed Abstract | CrossRef Full Text | Google Scholar
Depienne, C., Nava, C., Nava, B., Keren, S., Heide, A., Rastetter, S., et al. (2017). Genetic and phenotypic dissection of 1q43q44 microdeletion syndrome and neurodevelopmental phenotypes associated with mutations in ZBTB18 and HNRNPU. Hum. Genet. 136 (4), 463–479. doi:10.1007/s00439-017-1772-0
PubMed Abstract | CrossRef Full Text | Google Scholar
Eble, T. N., Sutton, V. R., Sangi-Haghpeykar, H., Wang, X., Jin, W., Lewis, R. A., et al. (2009). Non-random X chromosome inactivation in Aicardi syndrome. Hum. Genet. 125 (2), 211–216. doi:10.1007/s00439-008-0615-4
PubMed Abstract | CrossRef Full Text | Google Scholar
Edwards, T. J., Sherr, E. H., Richards, A. J. L. J., and Richards, L. J. (2014). Clinical, genetic and imaging findings identify new causes for corpus callosum development syndromes. Brain 137 (6), 1579–1613. doi:10.1093/brain/awt358
PubMed Abstract | CrossRef Full Text | Google Scholar
Elson, E., Perveen, R., Donnai, D., Wall, S., and Black, G. C. (2002). De novo GLI3 mutation in acrocallosal syndrome: Broadening the phenotypic spectrum of GLI3 defects and overlap with murine models. J. Med. Genet. 39 (11), 804–806. doi:10.1136/jmg.39.11.804
PubMed Abstract | CrossRef Full Text | Google Scholar
Fang, Q., Benedetti, A. F., Ma, Q., Gregory, L., Li, J. Z., Dattani, M., et al. (2016). HESX1mutations in patients with congenital hypopituitarism: Variable phenotypes with the same genotype. Clin. Endocrinol. 85 (3), 408–414. doi:10.1111/cen.13067
PubMed Abstract | CrossRef Full Text | Google Scholar
Fischer-Zirnsak, B., Segebrecht, L., Schubach, M., Charles, P., Alderman, E., Brown, K., et al. (2019). Haploinsufficiency of the Notch ligand DLL1 causes variable neurodevelopmental disorders. Am. J. Hum. Genet. 105 (3), 631–639. doi:10.1016/j.ajhg.2019.07.002
PubMed Abstract | CrossRef Full Text | Google Scholar
Flores, B., Delpire, C. C. E., and Delpire, E. (2019). A role for KCC3 in maintaining cell volume of peripheral nerve fibers. Neurochem. Int. 123, 114–124. doi:10.1016/j.neuint.2018.01.009
PubMed Abstract | CrossRef Full Text | Google Scholar
Franz, H., Villarreal, A., Heidrich, S., Videm, P., Kilpert, F., Mestres, I., et al. (2019). DOT1L promotes progenitor proliferation and primes neuronal layer identity in the developing cerebral cortex. Nucleic Acids Res. 47 (1), 168–183. doi:10.1093/nar/gky953
PubMed Abstract | CrossRef Full Text | Google Scholar
Friocourt, G., Poirier, K., Rakić, S., Chelly, J. G. J., and Chelly, J. (2006). The role of ARX in cortical development. Eur. J. Neurosci. 23 (4), 869–876. doi:10.1111/j.1460-9568.2006.04629.x
留言 (0)