
記住我


Liver transplantation is the only treatment available for pediatrics with end-stage liver disease. However, brain injury is a classic problem after pediatric liver transplantation [1]. Children suffer from neurological problems after liver transplantation in up to 46% of cases, with a higher mortality rate in the pediatric age group compared with adults [2]. Its symptoms are insidious and not easily detected, and may have long-term effects on children’s intellectual development.
Clinical studies have shown that the application of general anesthesia in infants and children under the age of 2 years leads to behavioral changes over a longer period of time, suggesting that general anesthetics may damage the central nervous system in infants and children [3]. But most of the children undergoing this type of surgery are at a sensitive stage of neurological development. The neuronal proliferation, differentiation, and migration as well as synaptogenesis, modification, and myelin formation in the brain are very active, and neural development is exceptionally sensitive to changes in the internal and external environment at this period of time [4]. Ferroptosis is a newly discovered mode of cell death in recent years, characterized by iron-mediated accumulation of lipid peroxides, which is associated with several oxidative stress diseases. The main iron storage places in the body are the liver, spleen, and macrophages. Both the liver and macrophages are important members involved in hepatic ischemia-reperfusion (HIR). Sevoflurane is widely used as an inhalation anesthetic in pediatric surgical clinical anesthesia [5]. However, recent studies have found that sevoflurane may be able to cause widespread neurotoxicity [6], and to date, there has been little agreement on the effects of sevoflurane in postoperative brain injury in the clinic. The relationship between ferroptosis and brain damage and sevoflurane has not been reported. Therefore, the present study was proposed to evaluate the effect of sevoflurane in brain injury after HIR in young rats and its relationship with ferroptosis.
Methods Animals and animal groupsSixty-four male Sprague-Dawley rats (2 weeks), weighing 20–30 g. All animals were brought from the Institute of Medical Laboratory Animals at the Chinese Academy of Medical Sciences and were kept in the same unit in a temperature-controlled environment [(22 ± 1) °C]. The rats were fasted for 12 h before the experiment and drank water freely.
After being numbered according to body weight, the rats were randomly divided into four groups using the random number table. The number of rats in each group was 16. The experimental groups were as follows: sham-operated group (S group, n = 16), the model group receiving HIR (HIR group, n = 16), sevoflurane group treated (HIR+Sev group, n = 16), and desferrioxamine treated group [deferoxamine (HIR+Sev+DFO) group, n = 16]. In HIR+Sev and HIR+Sev+DFO groups, rats were placed in an anesthetizing chamber and exposed to 3.6% sevoflurane [7] (Cayman, 23996, USA) with complete oxygen for 2 h, and sham and HIR group rats were conducted with the same procedure without sevoflurane exposure. DFO (100 mg/kg, MCE, HY-B0988, China) was administered continuously daily for 6 days before surgery in the HIR+Sev+DFO group [8]. Other groups were given equal amounts of saline.
Preparation of rat liver transplantation modelWe established a 70% HIR model following the steps described by Yu et al. [9] in the HIR, HIR+Sev, and HIR+Sev+DFO groups. Intraperitoneal injection of 3% sodium pentobarbital (30 mg/kg, glpbio, GC18059-10, USA) was used as preoperative anesthesia. After satisfactory anesthesia, the mice were fixed in a supine position, the skin was prepared and disinfected, the upper abdomen was opened along the abdominal midline to expose the hepatic hilum, and the left hepatic artery and portal vein were blocked with vascular forceps for 1 h, leaving the middle and left lobes of the mice in an earthen ischemic state, accounting for approximately 70% of the total mouse liver. Warm saline gauze was used to cover the wound. Then the vascular clamp was removed and the skin incision was sutured layer by layer. Rectal temperature was continuously monitored during surgery. A heat lamp was used to maintain the body temperature at about 37 °C. The sham group was separated from the vessels and bile duct tips without blocking the vessels.
After 6 h of reperfusion, the rats were sacrificed, eight rats were taken, Blood samples were obtained from the inferior vena cava, and serum was extracted by centrifugation at 3000 ×g for 15 min. Then each heart was opened along the right auricle after opening the thorax, 50 ml of prechilled heparin saline was perfused from the aorta through the heart, the head was quickly severed, and the intact brain was removed from the severed head, and hippocampal tissues and cortical tissues were picked. The hippocampal tissues and cortical tissues were stored in a refrigerator at −80 °C for the next step of detection; the other four rats were perfused with electron microscopic fluid using the above method, and the brain tissues were taken and fixed using 2.5% glutaraldehyde solution (Merck, 354400, Germany); the other four rats were perfused with saline as described above, and the brain tissues were taken and fixed using 4% paraformaldehyde solution(Merck, V900894, Germany).
General histologySections of brain tissue (five slices per sample) were collected, fixed, dehydrated, and transparently rendered in ethanol and xylene solution. The wax-soaked tissue is embedded in the embedding machine. Place the trimmed wax block cool at −20 °C freezing table, and slice the modified tissue chip wax block on the paraffin slicer(Leica RM2016 Shanghai, China), the slice thickness is 4 μm. Stain sections with Hematoxylin (HE dye solution set, Servicebio, Wuhan, China) solution for 3–5 min, and rinse with tap water. Then treat the section with hematoxylin differentiation solution, and rinse with tap water. Treat the section with Hematoxylin Scott Tap Bluing, and rinse with tap water. A total of 85% ethanol (SUPELCO, 1009832500) for 5 min; 95% ethanol for 5 min; finally stain sections with eosin dye for 5 min. Photomicrographs were captured with a light microscope (Nikon Eclipse E100, Tokyo, Japan). The nucleus was blue-purple, and the cytoplasm was red.
To calculate the number of abnormal neurons, three brain slices were randomly selected, and five nonoverlapping high-power fields (×400) in the hippocampal CA1 sector were randomly selected in each slice. Then, the number of abnormal neurons in the regions was calculated. By calculating the average number of abnormal neurons, statistical analysis was acquired for each group of samples.
Transmission electron microscope observation of hippocampusPrepare the desired samples as described by Zuo et al. [10]. Briefly, rats were prechilled by aortic perfusion with PBS (Cellgro, 21-040-CV). Complete hippocampal bodies were quickly removed, and the middle portion was selected and cut into small pieces (1 × 1 × 1 mm3). They were fixed with 2.5% glutaraldehyde for 2 h, rinsed with PBS, fixed with 1% osmium tetroxide (Honeywell, 201030) for 2 h, dehydrated with graded alcohol, embedded with Epon, and stained with lead citrate(Cayman, 701041) and uranyl acetate(Fluka, 73943-5G). Finally, the samples were observed by transmission electron microscopy (Hitachi HT7700, Japan). Two sections were selected for TEM observation, and five high magnification (×6000) images were randomly selected for each section.
Western blotProteins were extracted from freshly frozen tissues. Briefly, the tissues were homogenized and lysed with radio immunoprecipitation lysis buffer (Solarbio, R0010, Beijing, China), which has 100 mg/ml of phenylmethanesulfonyl fluoride (ADOOQ, A11901, California, USA) and 1 mg/ml of Aprotinin (ADOOQ, A14263). The lysate was collected and centrifugated for liquid supernatant. A total of 50 μg protein samples were separated with 10% SDS-PAGE (Solarbio, P1040) and transferred onto polyvinylidene fluoride (Millipore, HVLP04700, Arklow, Ireland) membranes. Membranes were blocked at room temperature with 5% skim milk (ACMEC, AC11037, Shanghai, China) powder in TBS (Quartett, 402000192, Berlin, Germany) for 1 h. Membranes were then incubated overnight at 4 °C with primary antibodies against the following proteins: Acyl-CoA synthetase long-chain familymember 4 (ACSL4) (1: 1000, ABclonal, A6826, Wuhan, China), transferrin receptor (TFRC) (1: 1000, ABclonal, A5865, Wuhan, China). Secondary antibodies (1: 1000, ABclonal, AS014, Wuhan, China) were incubated for 1 h at 37 °C and washed three times with PBST (PBS+0.1% Tween 20) (Biomed, PA202, Beijing, China). The protein bands were visualized by Molecular Imager Gel Doc XR System (Bio-Rad, Hertfordshire, UK) and quantified by densitometric analysis using an image analyzer (NIH Image J software, Bethesda, USA).
Enzyme-linked immunosorbent assaysBlood samples for analysis of S100β and neuron-specific enolase (NSE) were collected from the femoral vein, Serum S100β and NSE concentrations were detected by using the corresponding enzyme-linked immunosorbent assay kit (Shanghai mlBio Corp, ml003063, Shanghai, China; Beijing GERSION Corp, QS41610, Beijing, China) following the manufacturer instructions.
Statistical analysisGraphpad prism 6.0 statistical software (GraphPad Software, San Diego, California, USA) and SPSS 20.0 (IBM SPSS software, Armonk, New York, USA) were used for statistical analysis and graphing, respectively. All data are expressed as the mean ± SEM. Comparisons between the study groups were done using a one-way analysis of variance test. Post hoc analysis was done using Bonferroni’s method. A probability level of P < 0.05 was considered statistically significant.
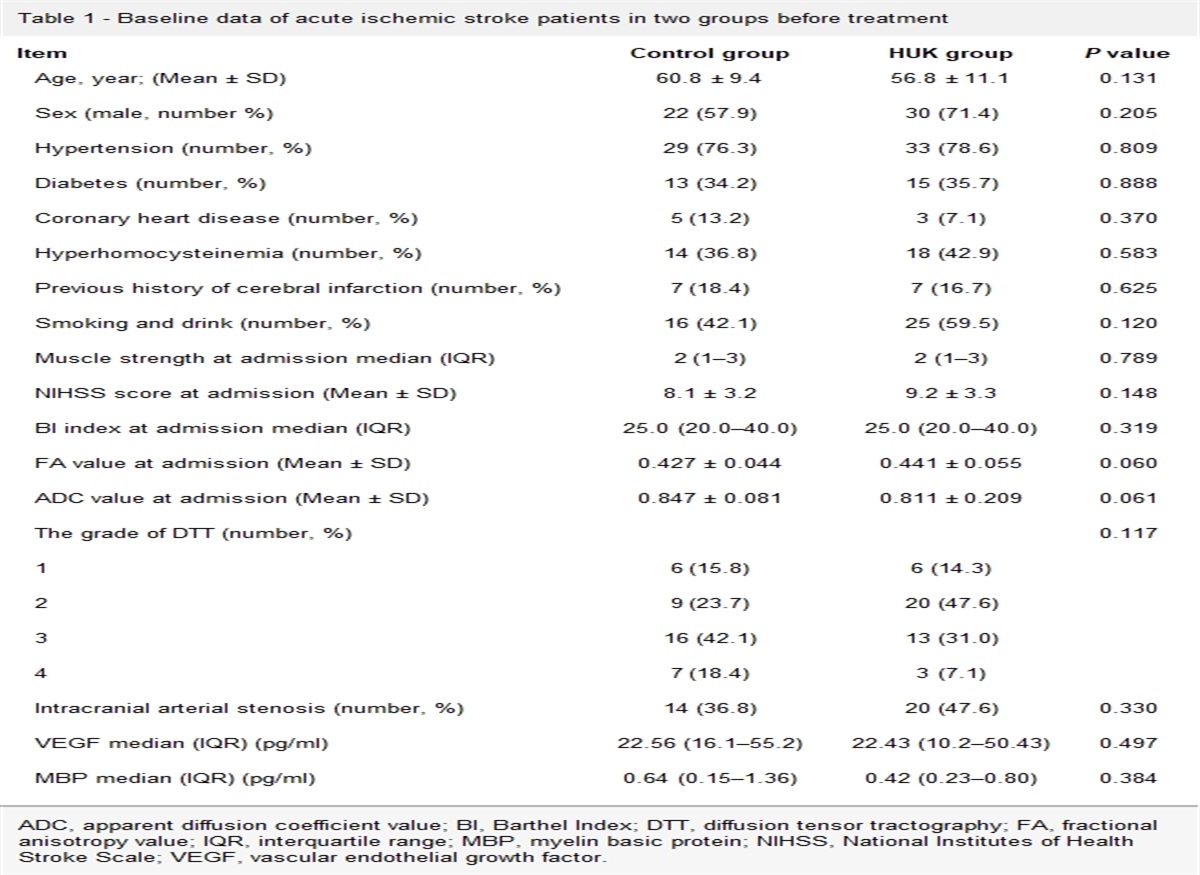
ResultsIn this study, we observed postoperative morphological changes of neurons in rat’s hippocampal CA1 region by HE staining (Fig. 1a). As shown in Fig. 1a, we found the neurons in the sham group were round or oval with rich, large, and lightly stained cytoplasm, and orderly arranged nuclei, significant histological abnormalities were seen in the HIR-induced group, showing single neuronal crinkling, deepened cell staining, and poorly demarcated nuclei and cytoplasm (black arrows). In contrast, in the 3.6% sevoflurane-treated group, multiple neuronal crinkling and increased damage were seen in the visual field, disorderly arranged nuclei, and neuronal pyknosis in the hippocampus were evident in this group. Compared with the sevoflurane group, the morphological pattern of hippocampal sections in the 100 mg/kg dose DFO-treated group showed reversal and improvement, the hippocampal neurons of rats in this group showed normal morphology, similar to the sham group.
 Fig. 1:
Fig. 1: Morphological changes of brain tissues in each group (HE staining,×200,×400; scale bars 100 and 50 μm) (a), quantitative analysis of injured neurons with neuronal pyknosis in the hippocampal CA1 region (b). Data were represented as the mean ± SD (n = 4, per group). ***P < 0.001 vs. the sham group. ###P < 0.001 vs. the HIR group. &&&P < 0.001 vs. the HIR+Sev group.
The number of damaged neuronal cells (Fig. 1b) in the HIR group was significantly higher than that in the S group, and the increase in neuronal damage was more pronounced in the HIR+Sev group; neuronal damage in the HIR+Sev+DFO group was obviously decreased and lower than that in the HIR group, but still higher than that in the S group (P < 0.001). It indicates that rats’ postoperative cognitive function may be related to the morphological changes of hippocampal neurons, and DFO can effectively alleviate the neural damage in the hippocampal CA1 region of the brain due to surgery and sevoflurane.
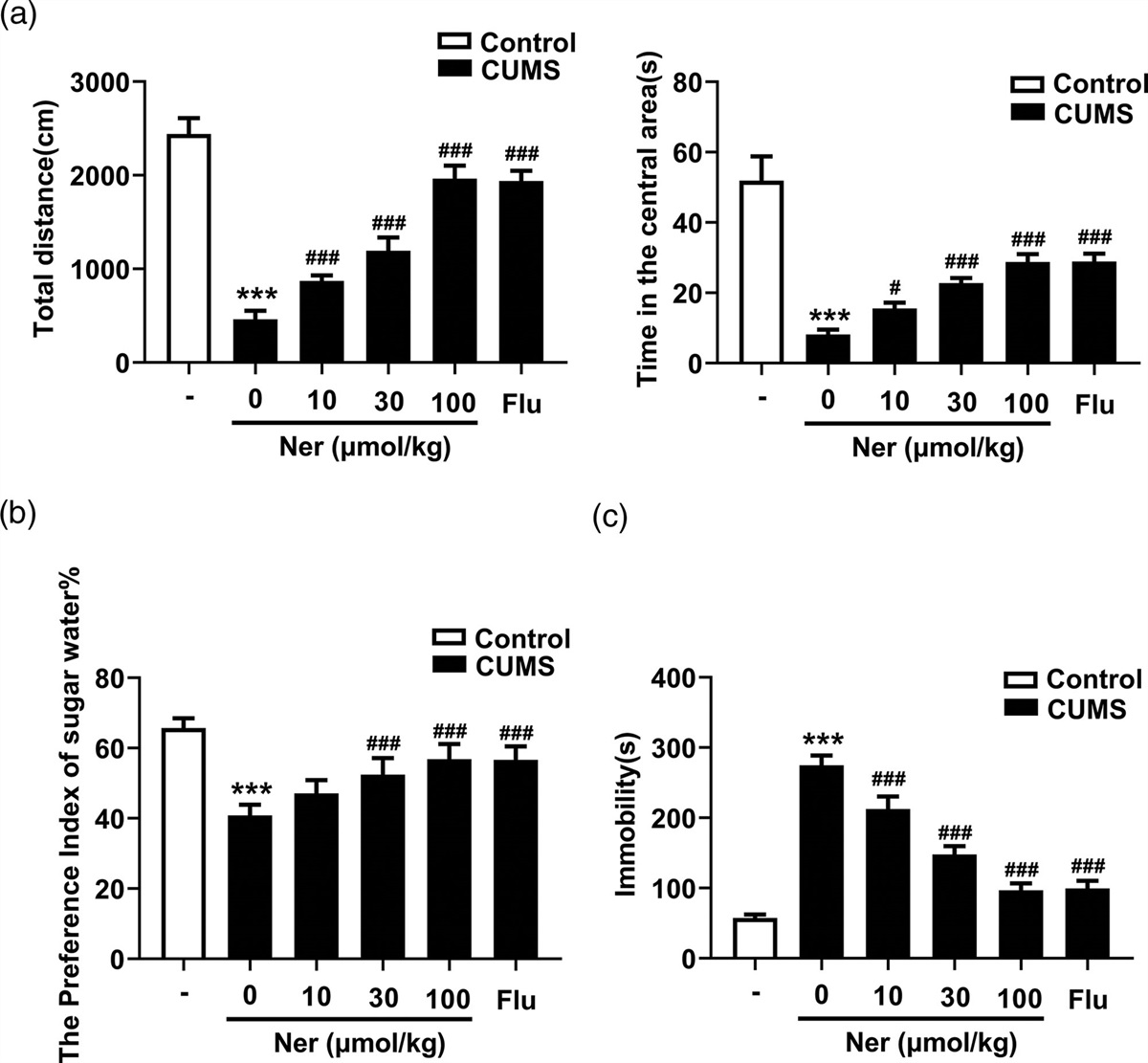
Iron inhibitors attenuate mitochondrial structural changes in hippocampal neurons caused by HIR and sevofluraneTo investigate the characteristics of ferroptosis in the hippocampal region after HIR, we examined the ultrastructure of neurons in the hippocampal region. As shown in Fig. 2, we found that in the HIR group, mitochondrial cristae were reduced or disappeared, and the same cristae reduction or disappearance was observed in the sevoflurane group. The situation was improved in the HIR+Sev+DFO group compared with the sevoflurane group. Our results showed that both surgery and sevoflurane can cause mitochondrial ferroptosis in neurons in the hippocampal region of the rat brain, and HIR+Sev+DFO can alleviate the altered mitochondrial ferroptosis in neurons.
 Fig. 2:
Fig. 2: Ultrastructure of hippocampal neurons in young rats of each group. (magnification×1500 and 6000; scale bars 5.0 and 1.0 μm). Black arrows indicate normal mitochondria; red arrows indicate reduced mitochondrial cristae.
Sevoflurane exacerbates the increase in ACSL4 and TFRC caused by Hepatic IR; however, this can be rescued by ferroptosis inhibitor DFO.
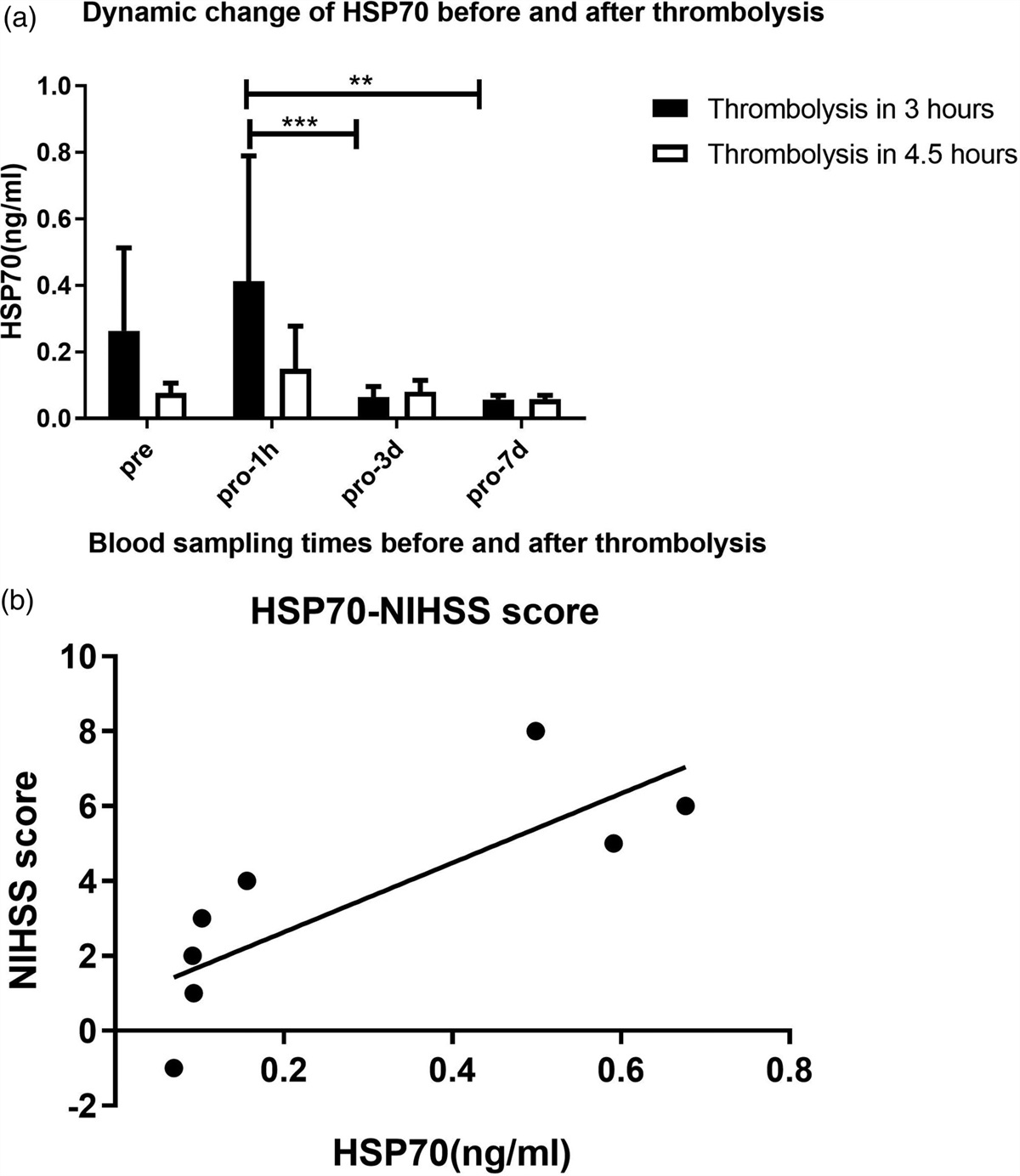
To determine whether ferroptosis participate in the brain of young rats after HIR, ACSL4 and TFRC were measured using western blotting (Fig. 3a). ACSL4 is an indicator of lipid peroxidation, whereas TFRC is an indicator of iron metabolism. In the hippocampal region of the juvenile rat brain, TFRC and ACSL4 levels in the HIR group were higher than those in the S group, and the elevation was more pronounced in the HIR+Sev group; the levels in the HIR+Sev+DFO group were markedly decreased and lower than those in the HIR and HIR+Sev group (P < 0.001), and there was no apparent difference in the levels compared with the S group (P > 0.05) (Fig. 3b and c). The trend of indicator expression was consistent in the cortex and hippocampus, and The content of ACSL4 in the cortical area of HIR+Sev+DFO was higher than that of the S group (P < 0.05) (Fig. 3c). Our findings demonstrated that surgery elevates ferroptosis indicators in both the cortex and hippocampus, that sevoflurane exacerbates ferroptosis, and that administration of the ferroptosis inhibitor DFO can markedly reduce the elevation of ferroptosis-related indicators.
 Fig. 3:
Fig. 3: Effect of Sevoflurane and DFO on ACSL4 and TFRC expression in hippocampus and cortex of brain after hepatic ischemia-reperfusion. Western blotting was used to measure the expression levels of ACSL4 and TFRC (a). Densitometry analysis of Western blots for the ratio of ACSL4 (b), TFRC (c). Data were represented as the mean ± SD (n = 8, per group). *P < 0.05 and ***P < 0.001 vs. the sham group. ###P < 0.001 vs. the HIR group. &&&P < 0.001 vs. the HIR+Sev group. ACSL4, Acyl-CoA synthetase long-chain familymember 4; TFRC, transferrin receptor.
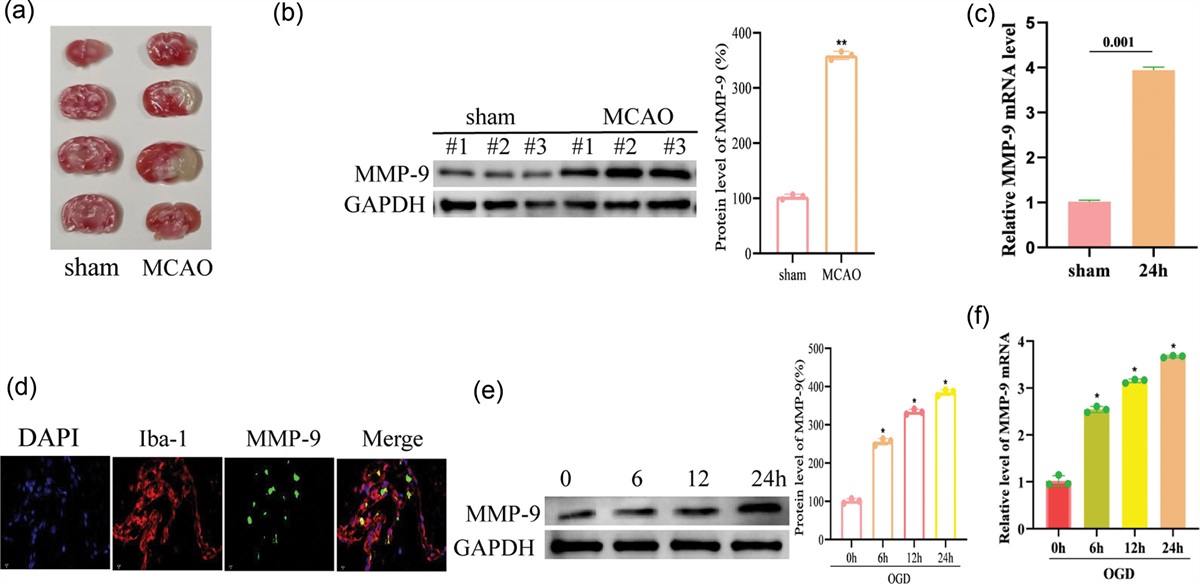
HIR and sevoflurane induced an increase in serum brain damage markersTo evaluate the degree of brain damage, the level of serum brain damage markers (NSE and S100β) was assessed. Compared with the S group, the concentrations of NSE in the hippocampus of rats increased significantly after surgery, and to a greater extent when sevoflurane was administered. The administration of DFO significantly reduced the concentrations of NSE, but their levels were still higher than those in the HIR group (P < 0.001) (Fig. 4a). Consistent with the trend of NSE, the concentration of S100β in the rat hippocampus increased significantly after surgery compared with the S group (P < 0.001), and the level in the HIR+Sev group increased compared with the HIR group (P < 0.001), and the concentration in the HIR+Sev+DFO group decreased compared with the HIR+Sev group (P < 0.05), but its level was still higher than that of the HIR group (P < 0.05) (Fig. 4b). This indicated that brain damage indicators in serum are elevated after surgery and sevoflurane aggravates the degree of brain damage, whereas administration of DFO effectively reduces brain damage indicators, but is still higher than in the HIR group.
 Fig. 4:
Fig. 4: HIR and Sevoflurane induced an increase in serum brain damage markers. The levels of serum brain damage markers: (a) NSE and (b) S100β. Data were represented as the mean ± SD (n = 8, per group). *P < 0.05 vs. the sham group. #P < 0.05 and ###P < 0.001 vs. the HIR group. &P < 0.05 and &&&P < 0.001 vs. the HIR+Sev group. NSE, neuron-specific enolase.
DiscussionIron is essential for the normal function of brain tissue, but iron-mediated oxidative stress is associated with the pathogenesis of ischemia-reperfusion injury [11]. Iron-mediated remote brain injury after HIR injury is rarely reported. In the present study, a 70% HIR young rat model was used. The results showed that after HIR, neuronal wrinkling, deepened cell staining, and indistinct cytoplasmic demarcation of the nucleus were seen in the region of the hippocampus of young rats, and the extent of such injury increased in the sevoflurane-treated group. Reduced or broken intracellular mitochondrial cristae of neurons in the hippocampal region were seen in both the HIR and sevoflurane-treated groups, ferroptosis signs were present. In contrast, ferroptosis inhibitor desferrioxamine effectively inhibited both HIR and sevoflurane-induced injury. The extent of ferroptosis was consistent with the extent of brain injury. It indicates that brain injury after HIR is associated with ferroptosis, and sevoflurane aggravates ferroptosis and brain injury in brain tissue after HIR in young rats.
Ferroptosis is a novel form of programmed cell death that is distinct from other forms of cell death and has iron-dependent characteristics [12]. Its essence is an iron-mediated impairment of lipid oxide metabolism, which causes cell death due to abnormal iron deposition inducing massive production of oxygen radicals, resulting in disruption of the balance of intracellular redox reactions. Ferroptosis mainly occurs in organs with active oxidative reactions, high energy metabolism, and rich in unsaturated fatty acids, especially in brain tissue [13,14]. Compared with the adult brain, the neonatal brain has a high rate of oxygen consumption, a high concentration of unsaturated fatty acids, and a low concentration of antioxidants, and the brain shows the lower activity of endogenous antioxidant defense mechanisms, which makes it particularly sensitive to oxidative damage [15]. Therefore, the young brain is more susceptible to ferroptosis-related damage.
If transferrin receptor 1, a receptor protein on the cell membrane sensitive to ferroptosis, is elevated, intracellular iron overload and excess Fe2+ will be combined with substances involved in redox reactions, such as hydrogen peroxide and superoxide radicals, to produce reactive oxygen species (ROS) through the Fenton reaction and Haber-Weiss reaction thereby inducing cell ferroptosis [16]. In the present study, we found that TFRC expression was elevated in the hippocampal region of young rats after HIR. Transferrin receptor is a cell membrane receptor that plays a key role in the regulation of cellular iron uptake. The results of the present study showed that transferrin receptor 1 was increased in brain tissue of remote organs after HIR, and the cellular uptake of iron was enhanced. Cells are in a state of iron overload. Ferroptosis plays a key role in the development of central nervous system disorders. It has been found that excessive iron accumulation in the brain will cause neurotoxicity and severe cognitive impairment [17]. The main role of TFRC is to assist transferrin (Tf) in the intracellular transport of iron ions. TFRC can inhibit the growth of neuronal protrusions [18]. Iron binding sites are found in both senile plaques and neurogenic fiber tangles, the amount of brain iron content is influenced by the amount of TFRC expression, and the removal of excess iron ions by desferrioxamine chelation can alleviate the symptoms of Alzheimer’s disease [19–21]. This may also contribute to the vulnerability to neurological disorders after liver transplantation.
ACSL4 is a member of the long-chain esteryl coenzyme A synthetase family. Recent studies showed that high ACSL4 expression rendered cells more sensitive to ferroptosis by preferentially catalyzing several polyunsaturated fatty acid such as arachidonic acid, and shaping cellular lipid composition [22] Alterations in ACSL4 enzyme activity can affect neurodevelopment through fatty acid metabolism [23]. ACSL4 can lead to ROS catalyzed by iron into lipid radicals, which produce cytotoxicity and trigger ferroptosis [20]. Ferroptosis has been associated with ischemic injury [24] and also with brain neuronal death in Parkinson’s disease [25]. ACSL4 can as well promote proinflammatory cytokine production of microglia [26]. Peroxidative stress in neurons can promote cognitive dysfunction through iron cell death. In the present study, we found that ACSL4 levels were elevated in the hippocampal region of young rats after HIR. This may be one of the main causes of remote organ damage after HIR.
Sevoflurane is a commonly used clinical anesthetic. Previous studies have shown that 1.5 MAC sevoflurane inhalation for 2 h potentiated surgery impaired cognitive function [27]. Prolonged (6–9 h) exposure to high concentrations (3–4%) of sevoflurane causes neuroinflammation and neuronal apoptosis and leads to long-term cognitive impairment [28]. Exposure to sevoflurane increased iron content in hippocampal neurons compared with cells not exposed to sevoflurane [29]. Sevoflurane enhances oxidative stress in the neonatal rat brains and induces ROS production and neuronal cell death [30]. However, it is unknown whether sevoflurane can affect brain injury after liver transplantation in young rats by modulating ferroptosis. In the present study, we found that TFRC levels and ACSL4 levels were elevated after sevoflurane administration, suggesting that ferroptosis occurs in whole-brain regions of young rats after HIR and that sevoflurane aggravates the extent of ferroptosis.
The iron chelator deferoxamine (DFO) has been used in animal model studies of various intracerebral iron overload diseases and neurodegenerative disorders [31]. Administration of desferrioxamine significantly inhibited the iron overload-induced increase in the generation of age spots, increased abnormal tau protein phosphorylation, and the resulting exacerbation of cognitive dysfunction in the brain of APP/PSL transgenic mice [32]. In the present study, we found that TFRC and ACSL4 levels were reduced in the brains of young mice after DFO administration, suggesting that DFO alleviated brain ferroptosis caused by HIR and sevoflurane. This is consistent with the findings of ferroptosis indicators and pathology. Moreover, the indicators of brain injury decreased after administration of DFO, suggesting that brain injury and brain ferroptosis are closely related.
The present study also has limitations, such as the small sample size of the rats selected, and the sample size needs to be expanded for further investigation in the future. In addition, it should be further investigated, which changes in intracellular ferroptosis cause neurological injury and whether mitochondrial cristae breakage is involved in neurological injury.
From the above discussion, the conclusion can be reached that sevoflurane increases brain injury after HIR by aggravating ferroptosis. Video abstract is available (see Supplementary Video, video abstraction.mp4, Supplemental Digital Content 1, https://links.lww.com/WNR/A676) for more insights from the authors.
AcknowledgementsThis work was supported by National Natural Science Foundation of China (No. 82072219) and the Youth Scientific Research Fund of the Second Hospital of Tianjin Medical University (No. 2020ydey08).
Guarantor of integrity of the entire study: X.Y. and X.Y.M. conceived the study and designed the experimental approach. X.Y., X.Y.M., J.S.L., Y.H.L., and K.K.W. conducted all experiments and data analyses. Y.H.L. and K.K.W. verified data analyses and performed data interpretation. X.Y. and X.Y.M. drafted the manuscript. Y.C.L., N.J., and W.L.Y. performed critical revisions to the draft. X.Y. and W.L.Y. prepared the final version for submission.
Our study was approved by the Ethics Review Board of Tianjin Medical University.
The data sets used and analyzed during the current study are available from the corresponding author upon reasonable request.
Conflicts of interestThere are no conflicts of interest.
References 1. Sun Y, Jia L, Yu H, Zhu M, Sheng M, Yu W. The effect of pediatric living donor liver transplantation on neurocognitive outcomes in children. Ann Transplant. 2019; 24:446–453. 2. Fernandez D, El-Azzabi TI, Jain V, Lloyd C, Wassmer E, Peake D, Gupte GL. Neurologic problems after pediatric liver transplantation and combined liver and bowel transplantations: a single tertiary centre experience. Transplantation. 2010; 90:319–324. 3. Brioni JD, Varughese S, Ahmed R, Bein B. A clinical review of inhalation anesthesia with sevoflurane: from early research to emerging topics. J Anesth. 2017; 31:764–778. 4. Krämer TJ, Sakas W, Jussen D, Krenzlin H, Kempski O, Alessandri B. Thrombin contributes to the injury development and neurological deficit after acute subdural hemorrhage in rats only in collaboration with additional blood-derived factors. BMC Neurosci. 2018; 19:81. 5. Liang LQ, Jiao YQ, Guo SL. Effects of sevoflurane inhalation anesthesia on cognitive and immune function in elderly patients after abdominal operation. Eur Rev Med Pharmacol Sci. 2018; 22:8932–8938. 6. Wang M, Zuo Y, Li X, Li Y, Thirupathi A, Yu P, et al. Effect of sevoflurane on iron homeostasis and toxicity in the brain of mice. Brain Res. 2021; 1757:147328. 7. Hu N, Wang C, Zheng Y, Ao J, Zhang C, Xie K, et al. The role of the Wnt/β-catenin-Annexin A1 pathway in the process of sevoflurane-induced cognitive dysfunction. J Neurochem. 2016; 137:240–252. 8. Pan K, Li X, Chen Y, Zhu D, Li Y, Tao G, Zuo Z. Deferoxamine pre-treatment protects against postoperative cognitive dysfunction of aged rats by depressing microglial activation via ameliorating iron accumulation in hippocampus. Neuropharmacology. 2016; 111:180–194. 9. Yu X, Jia L, Yu W, Du H. Dephosphorylation by calcineurin regulates translocation of dynamin-related protein 1 to mitochondria in hepatic ischemia reperfusion induced hippocampus injury in young mice. Brain Res. 2019; 1711:68–76. 10. Zuo W, Zhang S, Xia CY, Guo XF, He WB, Chen NH. Mitochondria autophagy is induced after hypoxic/ischemic stress in a Drp1 dependent manner: the role of inhibition of Drp1 in ischemic brain damage. Neuropharmacology. 2014; 86:103–115. 11. Scindia Y, Dey P, Thirunagari A, Liping H, Rosin DL, Floris M, et al. Hepcidin mitigates renal ischemia-reperfusion injury by modulating systemic iron homeostasis. J Am Soc Nephrol. 2015; 26:2800–2814. 12. Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci. 2016; 73:2195–2209. 13. Cruz-Alonso M, Fernandez B, Navarro A, Junceda S, Astudillo A, Pereiro R. Laser ablation ICP-MS for simultaneous quantitative imaging of iron and ferroportin in hippocampus of human brain tissues with Alzheimer’s disease. Talanta. 2019; 197:413–421. 14. Kushairi N, Phan CW, Sabaratnam V, David P, Naidu M. Lion’s mane mushroom, Hericium erinaceus (Bull.: Fr.) Pers. suppresses H2O2-induced oxidative damage and LPS-induced inflammation in HT22 hippocampal neurons and BV2 microglia. Antioxidants (Basel). 2019; 8:E261. 15. Wu Y, Song J, Wang Y, Wang X, Culmsee C, Zhu C. The potential role of ferroptosis in neonatal brain injury. Front Neurosci. 2019; 13:115. 16. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008; 15:234–245. 17. Masaldan S, Bush AI, Devos D, Rolland AS, Moreau C. Striking while the iron is hot: iron metabolism and ferroptosis in neurodegeneration. Free Radic Biol Med. 2019; 133:221–233. 18. Nakamura Y, Nakamichi N, Takarada T, Ogita K, Yoneda Y. Transferrin receptor-1 suppresses neurite outgrowth in neuroblastoma Neuro2A cells. Neurochem Int. 2012; 60:448–457. 19. Smith MA, Harris PL, Sayre LM, Perry G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci U S A. 1997; 94:9866–9868. 20. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014; 156:317–331. 21. Smith MA, Zhu X, Tabaton M, Liu G, McKeel DW Jr, Cohen ML, et al. Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J Alzheimers Dis. 2010; 19:363–372. 22. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017; 13:91–98. 23. Rodriguez JD, Bhat SS, Meloni I, Ladd S, Leslie ND, Doyne EO, et al. Intellectual disability, midface hypoplasia, facial hypotonia, and Alport syndrome are associated with a deletion in Xq22.3. Am J Med Genet A. 2010; 152A:713–717. 24. Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A. 2014; 111:16836–16841. 25. Do Van B, Gouel F, Jonneaux A, Timmerman K, Gelé P, Pétrault M, et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol Dis. 2016; 94:169–178. 26. Cui Y, Zhang Y, Zhao X, Shao L, Liu G, Sun C, et al. ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain Behav Immun. 2021; 93:312–321. 27. Hu N, Wang M, Xie K, Wang H, Wang C, Wang C, et al. Internalization of GluA2 and the underlying mechanisms of cognitive decline in aged rats following surgery and prolonged exposure to sevoflurane. Neurotoxicology. 2015; 49:94–103. 28. Luo A, Tang X, Zhao Y, Zhou Z, Yan J, Li S. General anesthetic-induced neurotoxicity in the immature brain: reevaluating the confounding factors in the preclinical studies. Biomed Res Int. 2020; 2020:7380172. 29. Wu J, Yang JJ, Cao Y, Li H, Zhao H, Yang S, Li K. Iron overload contributes to general anaesthesia-induced neurotoxicity and cognitive deficits. J Neuroinflammation. 2020; 17:110. 30. Fang H, Wang ZH, Bu YJ, Yuan ZJ, Wang GQ, Guo Y, et al. Repeated inhalation of sevoflurane inhibits the information transmission of Purkinje cells and delays motor development via the GABAA receptor ε subunit in neonatal mice. Mol Med Rep. 2018; 17:1083–1092. 31. Duscher D, Neofytou E, Wong VW, Maan ZN, Rennert RC, Inayathullah M, et al. Transdermal deferoxamine prevents pressure-induced diabetic ulcers. Proc Natl Acad Sci U S A. 2015; 112:94–99. 32. Tuo QZ, Lei P, Jackman KA, Li XL, Xiong H, Li XL, et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol Psychiatry. 2017; 22:1520–1530.
留言 (0)