
記住我
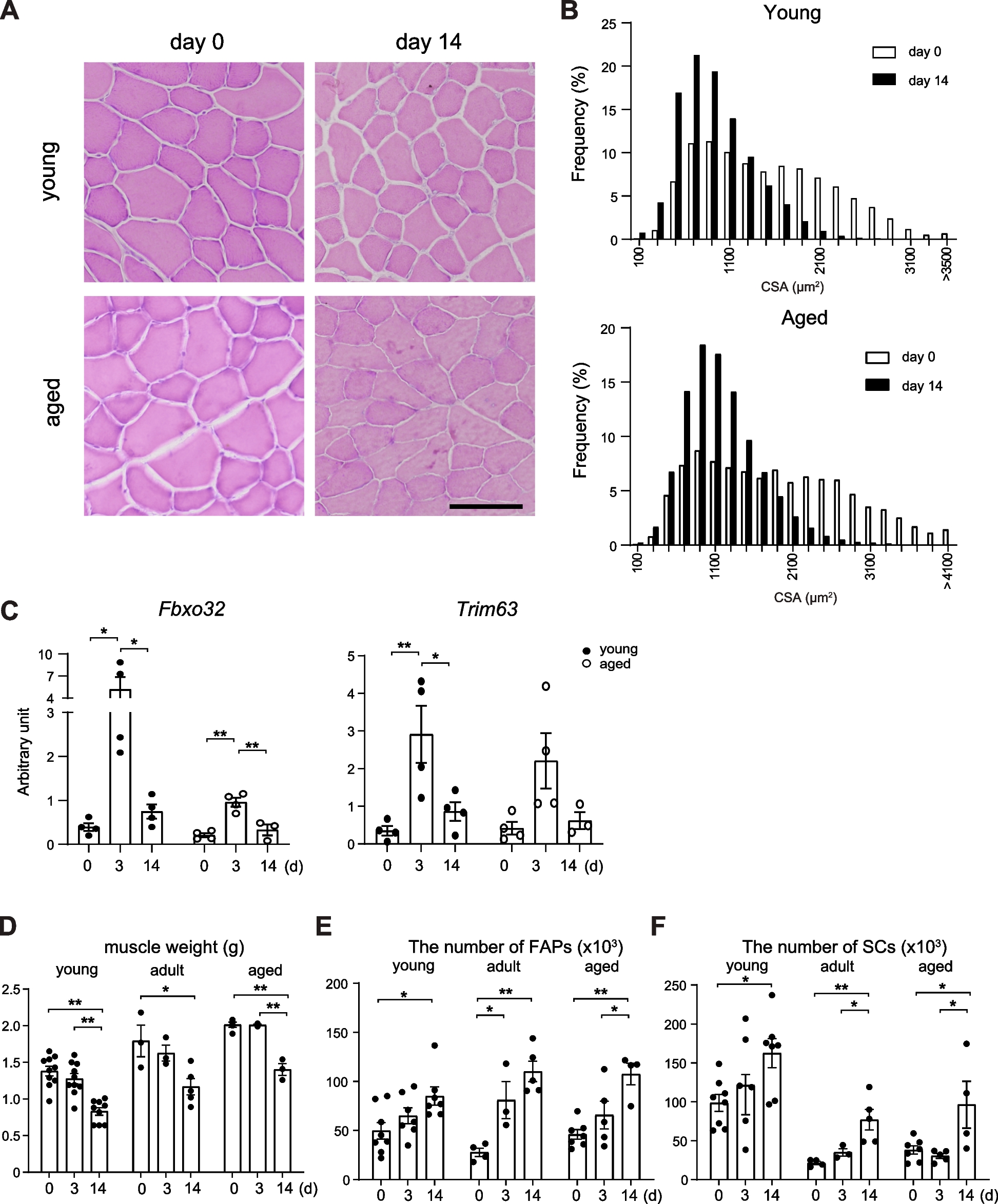





This study describes a new case of megaconial congenital muscular dystrophy (MCMD) due to two novel CHKB variants (Fig. 3A).
Fig. 3
Genotype–phenotype correlation in CHKB-mutated patients. A Scheme of CHKB gene (above) and the encoded choline kinase beta (bottom). Mutations identified in MCMD patients are indicated in red (group 1: non-sense/frameshift mutations), blue (group 2: missense/in-frame mutations) or black (mutations disclosed in our proband). B Distribution of age at onset (in years) in MCMD patients. C Relative comparison of best motor achievements in MCMD patients stratified according to genotype. D Prevalence of intellectual disability in MCMD patients stratified according to genotype. E Prevalence of heart disease (mainly dilated cardiomyopathy) in MCMD patients stratified according to genotype
MCMD has been described for the first time as a distinct muscle pathology in 1998 by Nishino and colleagues [1]. The genetic defect underlying MCMD was disclosed in 2011 with the identification of biallelic CHKB mutations in 15 MCMD patients displaying early-onset muscle weakness, mental retardation, and giant mitochondria at muscle biopsy analysis [2]. Phenotypic and histological overlap was observed with the spontaneous mutant rmd (“rostrocaudal muscular dystrophy”) mouse, which exhibited congenital muscular dystrophy and identical muscle fiber pathology [7, 8].
Although MCMD might display heterogenous multisystemic involvement, hypotonia and developmental delay without brain malformations associated to neuromuscular involvement characterized by slowly progressive proximal muscle weakness is the predominantly observed phenotype. Onset of symptoms is usually within the first 4 years of age (> 80% of cases described, Fig. 3B), but MCMD can also become manifest during adolescence. The cognitive involvement can resemble Rett syndrome [4] and early mortality is usually related to heart failure consequent to dilated cardiomyopathy. Ichthyotic skin changes, and seizures have been also frequently reported (30% of subjects). In few cases, worsening after stress/illness was reported [9,10,11].
So far, 45 patients from 40 independent families with documented CHKB mutations have been described (Supplementary Table 1) [3]. More than 60% of patients described are from Asia and 16 independent cases are of Turkish origin. At least 31 different mutations in CHKB were reported (Supplementary Table 1 and Fig. 3A). Most of them are nonsense mutations (8 stop codon and 8 frameshift variants), followed by missense (n = 10) and splice-site variants (n = 5). Thirty-five of 40 independent probands displayed homozygous defects supporting consanguinity in their unaffected parents.
We reviewed the clinical findings in patients reported so far (n = 45, Supplementary Table 1). Patients were divided into two groups based on their genotype. Group 1 included 33 patients whose pathogenic variants are expected to result in the formation of only truncated messenger RNA (mRNA), which is likely to be subject to nonsense-mediated decay (premature terminations codons, splice-site variants, or a combination of both). Group 2 included the remaining 12 patients whose variants are not expected to result in the formation of truncated mRNA (missense mutations or in-frame deletions).
There was a statistically significant difference (Mann–Whitney U test, p = 0.03662) in the mean age at onset between the two groups (group 1 2.00 ± 2.03 years, range from birth to 8 years of age, median age 12 months; group 2 6.80 ± 6.95 years, range from 1 month to 17 years of age, median age 3.5 years).
Mean age at walking was 2.45 ± 1.09 years for group 1 compared with 1.66 ± 0.70 years for group 2 (Mann–Whitney U test, p = 0.02574). Eight patients from group 1 (patients 6, 9, 11, 14, 17, 20, 29, and 30) never acquired independent ambulation [2, 4, 8, 12,13,14,15]. In group 2 unassisted ambulation was achieved in all patients with the exception of a single proband (patient 43) harboring the homozygous p.Glu283Lys change in the choline kinase motif of the enzyme [13].
Available data about the age at last examination showed a mean age of 10.77 ± 6.30 years for group 1 compared with 20.23 ± 12.27 years for group 2 (Mann–Whitney U test, p = 0.0271).
Considering the best motor skill gained, the proportion between “walkers” and “sitters” was 76% versus 24% in group 1 and 92% versus 8% in group 2 (Fig. 3C).
Cognitive impairment (autism spectrum disorder, attention deficit hyperactivity disorder, intellectual disabilities, limited intelligence) is present in virtually all the patients with the exceptions of two Canadian siblings (patients 38 and 39) who displayed initial symptoms in the second decade with mild disease course (Fig. 3D) [10]. Intellectual disabilities seem to be less severe in subjects from group 2. Group 1 patients present more frequently autistic features compared to group 2 (70% versus 45%, respectively).
Cardiac involvement (dilated cardiomyopathy, decreased left ventricular systolic function, congenital heart defects) has been reported in 11 of 33 group 1 MCMD patients (33%) and resulted fatal in six of them [6, 8, 9, 12,13,14,15]. Heart disease was reported in a single patient (patient 35) from group 2 in the considered observation time (Fig. 3E) [9].
Broad clinical heterogeneity, observed within each group, limit the range of this genotype–phenotype correlation. For example, 2 patients from group 1 displayed symptoms onset after 5 years of age (patients 7 and 13 [13]) and carried C-term truncating mutations expected to partially preserve the choline kinase catalytic site. Marked clinical heterogeneity was also reported in the same familial group as in the case of patient 6 (onset at 14 months of age and unable to walk) and his sibling patient 7 (onset after 6 years of age and walking independently at 3 years of age) [13].
In our patient, the p.Gly354Arg change affects a conserved residue downstream the choline kinase domain in the C-term region where other pathogenic variants were previously detected. The c.448-56del27 microdeletion acts a splicing modifier, resulting in a partial intronic retention leading to a shift in the reading frame at the level of the ATP binding loop. Therefore, our case displays a mixed genotype with a null allele in trans with a missense change. Age at onset and delayed age at walking seem to indicate that this genotype causes a severe form of MCMD resembling clinical presentations of group 1 patients. Intellectual disability was present, confirming this symptom as a cardinal feature of MCMD. So far, the patient has not developed cardiomyopathy at clinical or instrumental level, but cardiac monitoring is highly recommended.
Muscle biopsy examination was fundamental to drive molecular testing and provided a valuable resource for the validation of CHKB defects at transcript level and the execution of biochemical studies. MCMD features typical giant mitochondria with evidence of abnormal cristae at the periphery of the fiber at electron microscopy. Histological studies also showed enlarged mitochondria at the periphery of the fibers and rarefied areas at the center. Dystrophic features and mitochondrial changes are homogenously observed in all the muscle biopsies so far available from MCMD patients. Muscle deterioration is confirmed by increased (mild to moderate) serum CK levels (90% of tested patients).
The pathogenesis of MCMD, although not entirely understood, was extensively investigated in the last 15 years [3]. Investigations performed on Chkb-ko rmd mouse showed impaired respiratory function, increased reactive oxygen species, and enhanced mitophagy in muscle tissue [7, 8]. The introduction of a muscle-specific Chkb transgene fully rescued motor and behavioral function in the rmd mouse model, confirming the cell-autonomous nature of the disease. More importantly, AAV6-based intramuscular gene therapy improved dystrophy phenotype even after disease onset in preclinical models [16]. Altered lipid metabolism was also demonstrated to result in the increase of the arrhythmogenic lipid acylcarnitine predisposing to arrhythmia in hypertrophic cardiac muscles [17]. Tavasoli and colleagues have recently demonstrated that a temporal change in lipid metabolism occurs in Chkb − / − affected muscles. They observed that impaired β-oxidation of fatty acids in mitochondria results in triacylglycerol accumulation as the disease progresses. Interestingly, the decrease in peroxisome proliferator-activated receptors (PPAR) and downstream target gene expression can be reversed by pharmacological PPAR agonism [18].
Irregular mitochondrial morphology is linked to hampered mitochondrial fission consequent to decreased levels of the fission protein DRP1, compromising OXPHOS activity [19]. Aksu‐Menges and colleagues have recently observed altered mitochondrial morphology, reduced levels of mitochondrial fission proteins and derangement in several mitochondrial pathways in human primary skeletal muscle cells from a MCMD patient [20]. Our study confirms these findings in the muscle of our patient: engaged autophagy was indirectly suggested by increased levels of p62 and LC3 in some muscle fibers while decreased levels of DRP1 were associated with a severe multi-complex defect in presence of normal levels of respiratory chain protein subunits. The rarefication of mitochondria in the center of muscle fibers, observed in our case as well as in previous reports [1, 20], might be a consequence of sustained mitophagy.
Nowadays, modern diagnostic approach based on NGS sequencing bypass the need of invasive procedures to achieve a molecular diagnosis in a relevant number of patients with neuromuscular disorders. Nevertheless, we highlight the appropriateness of muscle biopsy for the validation of genetic findings and, as in the case of MCMD, for the identification of pathognomonic features which unequivocally direct the molecular analysis.
留言 (0)