
記住我
Genome-wide association studies (GWAS) of neurodevelopmental and psychiatric disorders have demonstrated that the majority of common variation associated with these disorders is found in noncoding regions of the genome [1,2,3,4,5,6,7,8]. Similarly, whole-genome sequencing studies (WGS) are poised to discover rare noncoding genetic variation associated with neurodevelopmental disorders [9,10,11]. Whereas the functional impact of genetic variation in protein coding regions can be inferred through knowledge of the codon code, the impact of genetic variation in the noncoding genome is much more difficult to understand as no such regulatory code is known. The noncoding genome contains cis-regulatory regulatory elements (CREs) such as enhancers, promoters, silencers, and insulators, which influence gene expression by serving as docking sites for DNA-binding proteins like transcription factors (TFs) [12, 13]. Variants within a regulatory element can alter TF binding and subsequently alter gene expression and cellular function [14, 15].
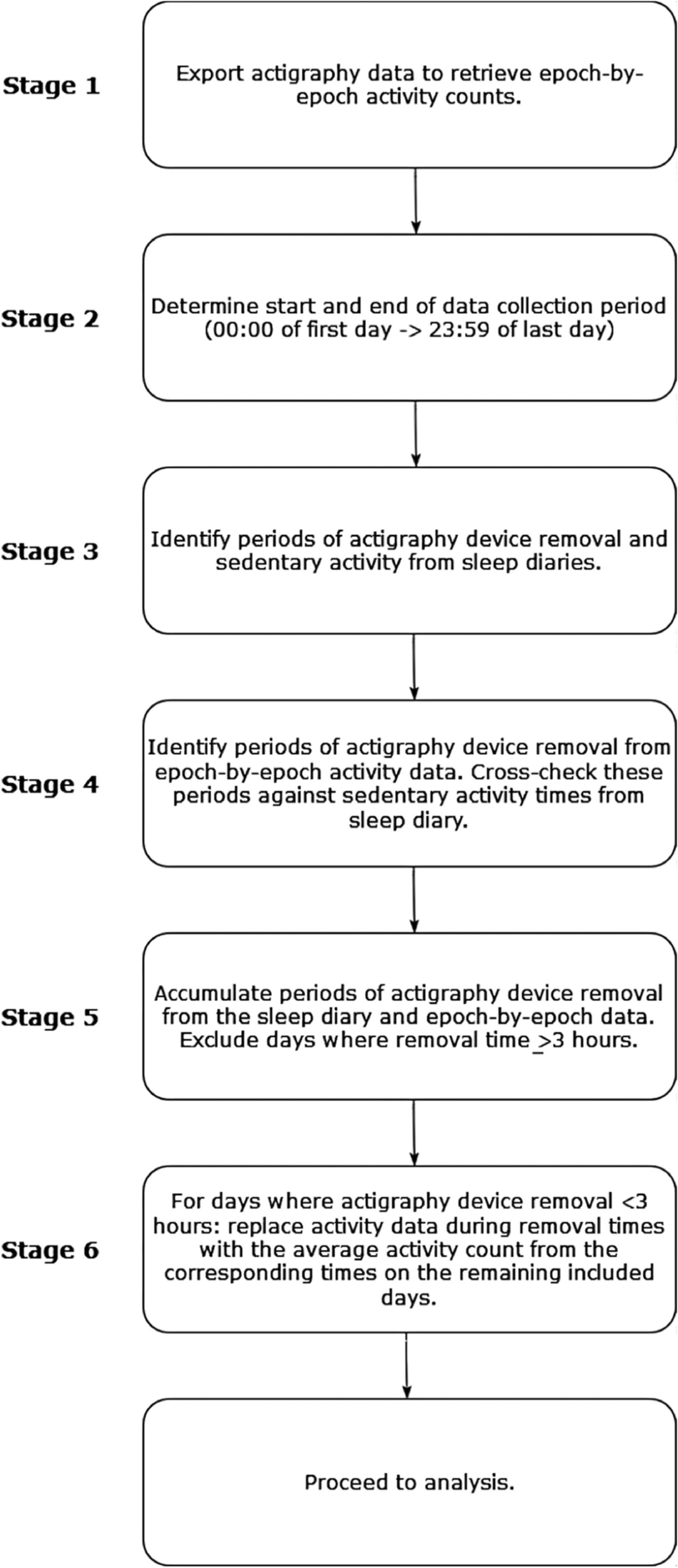
In addition to the lack of regulatory code, GWAS alone cannot pinpoint variants that are causing a disease because of linkage disequilibrium (LD), the nonrandom inheritance of nearby alleles on the genome. A genome-wide significant (GWS) locus typically contains tens to hundreds of single-nucleotide polymorphisms (SNPs) that are associated with a trait or disease. Only a subset of these SNPs are thought to be causal. While it is commonly thought that the index SNP, the SNP most significantly associated with the trait at a locus, is causal, growing evidence portrays a more complex picture [16]. The lead SNP is not always the causal allele when functionally validated, and a given locus can contain multiple causal variants [16]. Identifying the causal variant(s) at a locus can greatly facilitate our understanding of disease mechanisms by narrowing down the genetic underpinnings of a disease (Fig. 1). Moreover, causal variant identification provides the intriguing possibility of developing therapeutics by reversing pathological transcriptional mechanisms or genetically modifying causal variants [17, 18].
Fig. 1
Use of MPRA to identify causal variants at a GWAS locus containing many SNPs in high LD. A The schematic cartoon plots show GWAS and MPRA SNPs and their corresponding significance at a single locus. LD structure confounds identification of the causal variant in the GWAS, but the MPRA tests regulatory effects of each SNP independently so it can identify a specific causal variant. B. Top, SNP association statistics at a genome-wide significant locus from an ASD GWAS [19]. The index SNP, rs60527016, reached genome-wide significance. SNPs are colored by binned LD (r2) relative to the MPRA-validated variant (rs7001340). The existence of SNPs that are in high LD with rs7001340 highlights the difficulties in defining which SNPs are functional or causal based on GWAS alone. Bottom, MPRA identified a causal variant within this locus (rs7001340) that shows strong allelic regulatory activity. Image adapted from [19]
Several experimental and computational designs have been used to predict the causal variant at a given locus. Fine-mapping tools computationally predict potential causal variants based on association statistics and LD patterns [20,21,22,23], but different algorithms can yield conflicting results, and prioritized variants still require experimental validation [24]. Allele-specific chromatin accessibility (ASCA) can be used to determine if inherent genetic variation in a population of individuals affects chromatin accessibility, a proxy for gene regulatory activity, in relevant cell types [25, 26]. Genetic variants within a noncoding regulatory element that affects the function of that element are highly likely to be causal gene regulatory variants. Allele-specific chromatin accessibility when colocalized with GWAS suggests that the genetic variants are also causally associated with the trait or disease. However, ASCA experiments require large sample sizes of genetically diverse donors with both genotype and chromatin accessibility data, and they cannot independently test the effect of multiple variants in high LD within a regulatory element [27, 28].
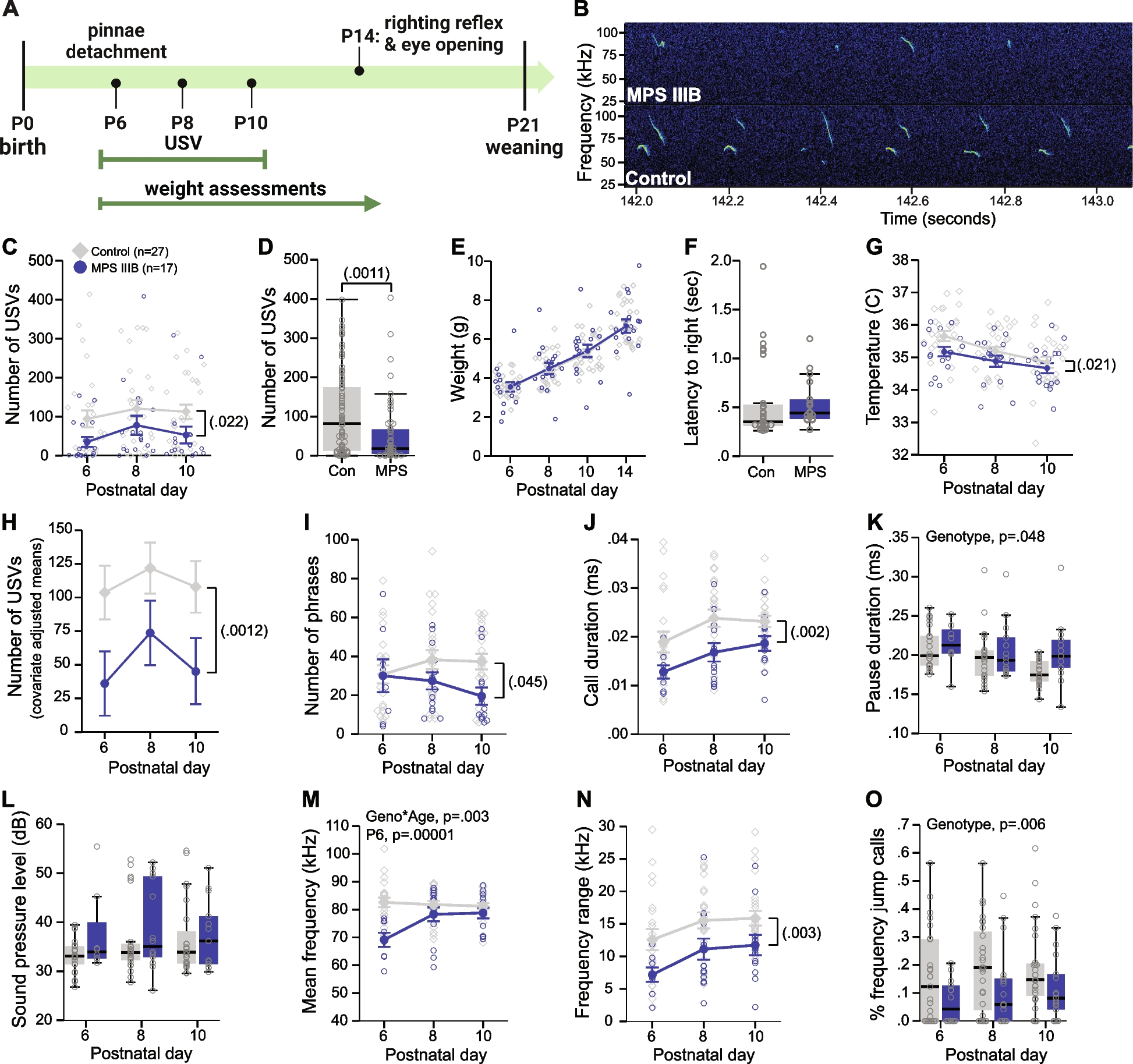
Functional validation assays fill the existing gap by experimentally demonstrating how genetic differences lead to phenotypic effects [29]. Gene regulatory activity of noncoding elements has historically been functionally validated via luciferase assays (Fig. 2A). A luciferase assay places a regulatory sequence of interest (sometimes containing a SNP) upstream of a luciferase reporter gene and quantifies the regulatory effect on expression via luminescence of luciferase [27]. However, luciferase assays lack the throughput to validate thousands of regulatory sequences at once because each regulatory element must be measured independently.
Fig. 2
Luciferase assay vs cis-regulatory MPRA. A Luciferase assay measures light emitted by a reporter gene, luciferase, driven by a regulatory element. B In a canonical cis-regulatory MPRA, the regulatory element drives the RNA expression of the unique barcodes. Transcriptional activity is quantified as barcode transcription (via RNA-seq of the barcodes) normalized to initial input of barcodes (via DNA-seq of the barcodes). Thousands of cis-regulatory elements (CREs) can be tested in the same experiment
Massively parallel reporter assays (MPRA) have advanced the throughput of luciferase assays by enabling the simultaneous functional validation of regulatory activity of thousands of variants on a massive scale (Fig. 2B), often vastly narrowing down thousands of variants found by GWAS or quantitative trait loci (QTLs) in a single assay (Fig. 1). Rather than quantifying the luminescence of luciferase, MPRA measures the barcoded reporter gene expression via next-generation sequencing. Once the MPRA construct is introduced into cells of interest, the synthesized regulatory element drives the expression of its unique barcode, a random oligo sequence that uniquely tags the matching regulatory element. The initial input of the construct is quantified by the DNA counts of the barcodes, which is compared to the RNA counts to evaluate effects on expression (Fig. 2B).
MPRA has incredible potential for studying the noncoding genetic variants associated with neurodevelopmental disorders. Whereas the majority of efforts have been made to characterize common variants identified by GWAS, wide application of WGS would further expand the utility of MPRA in characterizing various classes of variants located in the noncoding genome.
In this review, we will discuss the broad application of MPRA to functionally validate variants within the various regulatory contexts that encompass transcriptional and posttranscriptional regulation. We will then add important considerations for conducting MPRA, including limitations of MPRA experiments. We conclude by providing future directions of MPRA.
MPRA for studying cis-regulatory elementsCanonical MPRAThe canonical MPRA design includes a CRE, a generic promoter, a reporter gene, and a unique barcode assigned to each regulatory element (Fig. 2B). Generally, CRE libraries of interest are made with mass oligonucleotide synthesis. To interrogate variant effects on gene regulation, the CRE can be modified to harbor a variant within its sequence. Additionally, every possible single-nucleotide mutation can be added to the CRE, called saturation mutagenesis. The impact of the variant on regulatory activity is measured through barcodes matched to each unique variant. Because the barcode itself can have an influence on levels of expression, many barcodes are usually tested for each variant. Transcriptional activity is quantified as barcode transcription (via RNA-seq of the barcodes) normalized to initial input of barcodes (via DNA-seq of the barcodes). This allows systematic investigation of variant function within a noncoding region by comparing the gene regulatory activity between protective and risk alleles of a given variant. A growing body of research employs this strategy to identify functional regulatory variants within GWAS loci [30,31,32,33,34]. Key consideration in designing MPRA involves the use of proper controls [35]. For example, scrambled sequences of DNA in the relevant cell type can be used as negative controls to experimentally validate enhancers [35]. Likewise, a strong promoter or a known highly expressed sequence in the relevant cell type can be used as positive controls [35]. The canonical MPRA design has been adapted to fit the needs of differing types of CREs being tested [36,37,38,39].
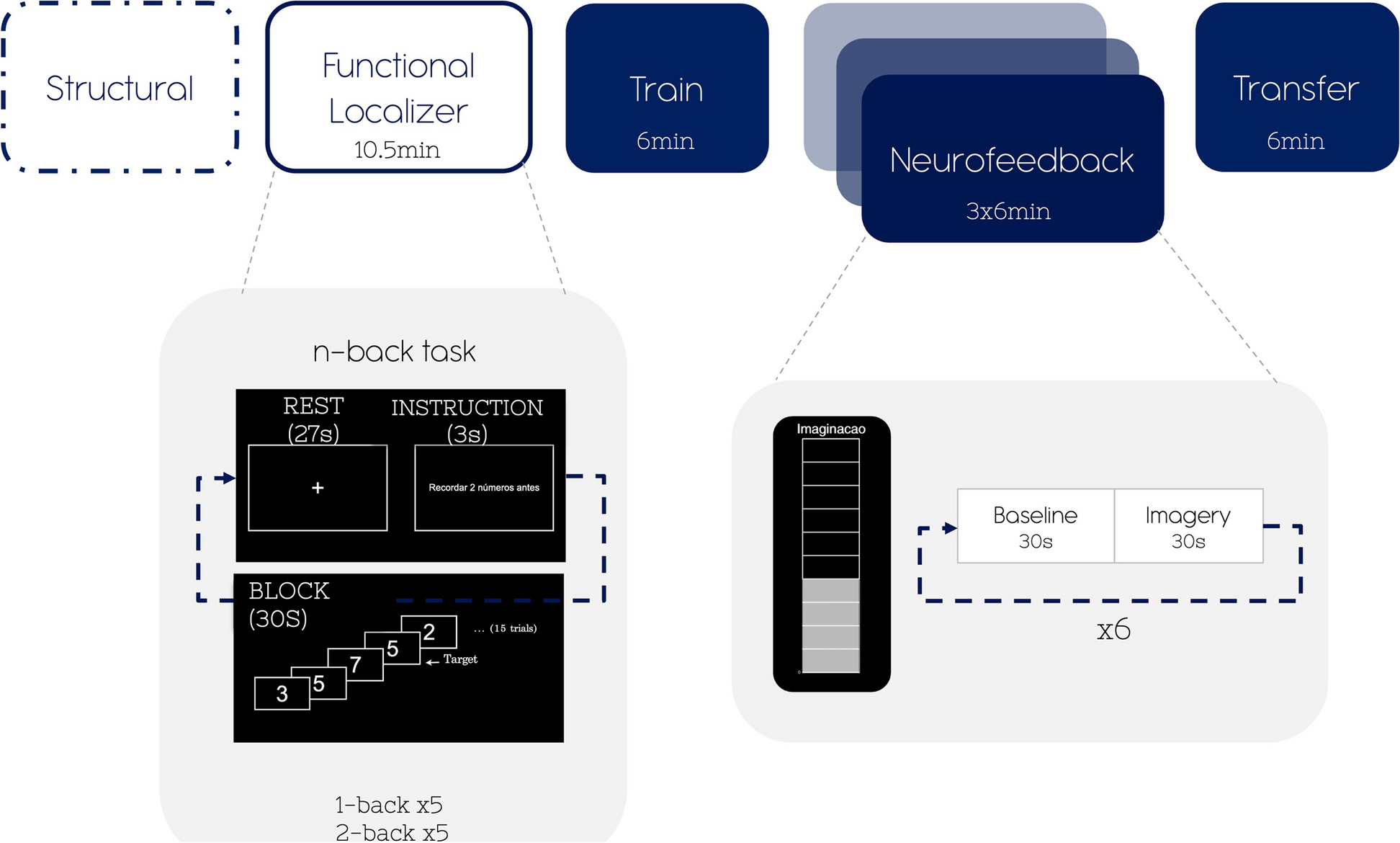
PromoterMutations and variants in promoter regions can have a profound impact on gene expression. MPRA has been used to test the impact of variants in promoters on a massive scale. In comparison with the canonical MPRA design, promoter MPRA lacks a CRE and alters the DNA sequence of the promoter region. Patwardhan et al. utilized promoter MPRA with saturation mutagenesis to screen the activity of mutated promoter sequences with attached barcodes (Fig. 3A) [40]. Barcode counts quantified via short-read sequencing provided a scalable readout of promoter activity, which led to the identification of critical regions of a promoter that govern transcriptional efficiency [40].
Fig. 3
MPRA designs for studying gene regulation. MPRA modifies the design of canonical cis-regulatory MPRA (described in Fig. 2B) that contains a cis-regulatory element (CRE), a promoter, a reporter gene, and a unique barcode (BC). Elements of this construct can be replaced or rearranged to test different types of CREs. The red vertical line indicates where a variant can be located. A Promoter MPRA contains a promoter harboring a variant, a reporter gene (e.g., GFP), and a unique BC. Image adapted from [40]. B Enhancer MPRA contains a regulatory element harboring a variant, a (minimal) promoter, a reporter gene, and a unique BC. C Transcription factor binding MPRA (TransMPRA) can be broken down into two components: (1) a promoter with a guide RNA (gRNA) that targets a transcription factor (TF) of interest and (2) a promoter, a test enhancer sequence harboring a variant, and a unique BC. The gRNA brings catalytically dead Cas9 protein with an attached Krüppel-associated box (dCas9-KRAB) which silences the expression of the TF gene. If the silenced TF interacts with the test enhancer, the downstream barcode expression is decreased. Image adapted from preprint [41]. D Silencer MPRA (in a STARR-seq style) contains a (strong) promoter and a test silencer harboring a variant. The silencer sequence can prevent self-transcription by silencing the promoter. Image adapted from [42]. E Splicing MPRA has minigene constructs that are inserted between a split-GFP reporter (GFP-N terminus and GFP-C terminus) and a peptide 2A (P2A) upstream of an mCherry reporter. Variants can be located in the variable intron sections on either side of the exon or within the exon. Inclusion of the middle exon disrupts GFP fluorescence, and cells can be FACS sorted into bins based on GFP:mCherry ratios. The GFP with or without the exon are quantified for exon inclusion or skipping via DNA-seq of the plasmid in each sorted bin. Image adapted from [43]. F RNA modification MPRA contains a promoter, an arbitrary coding sequence (CDS), a putative pseudouridine (Ψ) sequence as 3′ untranslated region (UTR), and a unique barcode. Once the library is introduced, cells are treated with N-cyclohexyl-N′-β-(4-methylmorpholinium) ethylcarbodiimide (CMC) which binds to Ψ and prevents reverse transcription (RT). High-throughput sequencing of cDNA allows prediction of the exact base pair location of the Ψ RNA modification. Variants can be inserted anywhere in the CDS. Image adapted from [44]. G 3′ UTR MPRA consists of a promoter, a reporter gene, a 3′ UTR harboring a variant, and a BC. BC RNA counts reflect transcriptional stability modulated by 3′ UTRs. H RNA localization MPRA consists of a promoter, a mutated Sox2 gene that localizes in the cytoplasm (fsSox2), a lncRNA harboring a variant, and a unique barcode. Barcode expression from subcellular fractions is used to interrogate subcellular localization of lncRNA. Image adapted from [45]
In addition to introducing variation in the promoter sequences, a similar approach can be used to characterize promoter activity of any given sequence. Boer et al. developed a gigantic parallel reporter assay (GPRA) that measured the promoter activity of over 100 million randomly synthesized sequences [46]. The complexity of synthetic promoters surpasses the complexity of the human genome, allowing them to build a predictive model of how genetic sequence affects transcriptional regulation.
While many neurodevelopmental disorder-associated variants have been shown to be enriched in promoter regions [24, 47, 48], MPRA has yet to be adopted to systematically examine the regulatory function of these promoter variants. We expect MPRA will provide a useful avenue to elucidate the function of promoter variants associated with neurodevelopmental disorders.
EnhancersEnhancers are CREs that TFs bind to and activate gene expression [49]. Disease-associated risk variants are enriched in enhancers [50]. Despite their important roles in gene regulation and disease associations, the sequence logic of enhancers is not well understood. Therefore, MPRA has been widely adapted to experimentally test the function of enhancers and variants within enhancers [19, 51]. While MPRA can take on many forms to examine enhancer functions [52], generally, a putative enhancer element is coupled with a weak promoter (e.g., minimal promoter) that is followed by a reporter gene and a unique barcode (Fig. 3B).
Myint et al. used enhancer MPRA to screen 1049 schizophrenia- and 30 Alzheimer’s disease-associated variants for differences in driving reporter gene expression [34]. They used two cell lines and identified 192 SNPs with significant differences in driving reporter gene expression [34]. Among the 192 variants, 148 showed allelic differences in K562 cells, 53 in SK-SY5Y cells, and only 9 showed allelic differences in both cell lines, demonstrating that genetic variants often exert their regulatory effects only within specific cell types [34]. As an additional example, Matoba et al. used MPRA to fine-map one novel ASD GWAS locus in HEK293T cells (Fig. 1) [19]. Of 98 variants tested, two were found to have significant differential allelic activity, with one variant (rs7001340) exhibiting strong effects. By integrating MPRA results with expression quantitative trait loci (eQTLs), they showed that an ASD-associated risk allele decreased the expression of DDHD2 [19]. These examples highlight MPRA’s ability to map disease-associated variants within putative enhancer regions.
Transcription factors (TF) recognize and bind to specific sequences within an enhancer, called TF binding motifs, to regulate gene expression. Variants within motifs can disrupt TF binding or create new motifs, altering regulatory activity. Though enhancer MPRA can identify if a variant affects enhancer activity, it does not experimentally validate which TF contributes to the altered regulation. TF-DNA interactions can be measured using methods such as chromatin immunoprecipitation sequencing (ChIP-seq), or they can be inferred using CRISPR knockout screens that model the impact of TFs on gene regulatory programs [53,54,55]. A recently introduced technique (in preprint) called TransMPRA also addresses this question by combining MPRA with CRISPR interference and single-cell sequencing to measure the interaction between transacting factors and putative enhancers (Fig. 3C) [41]. In this system, a guide RNA (gRNA) for a known TF is packaged together with enhancer MPRA that are potentially directly targeted by the TF [41]. When introduced into cells expressing dCas9-KRAB proteins, TF expression is inhibited, and enhancer activity is reduced only if the element is a downstream target of the TF [41]. Accordingly, TransMPRA provides an incredibly important tool to delineate potential transcriptional regulators for noncoding variants associated with neurodevelopmental disorders.
SilencersMPRA has been adapted to test silencer elements, which are noncoding functional elements that lead to decreased expression of their target gene (Fig. 3D) [42]. Silencer MPRA differs from enhancer MPRA in two aspects. First, enhancer MPRA uses a weak promoter (e.g., minimal promoter) to measure increases in gene expression elicited by the putative enhancer, while silencer MPRA uses a strong promoter (e.g., super core promoter, SCP1) that transcribes a high baseline level of the construct, so decreases in transcription can be detected. Second, silencer MPRA leverages the design of self-transcribing active regulatory region sequencing (STARR-seq), a sub-branch of MPRA (for more information about STARR-seq, please see the review [56]). STARR-seq places an uncharacterized CRE downstream of a strong core promoter followed by a polyA tail. This MPRA design does not require barcodes because the sequence of the transcribed putative silencer acts as the barcode [42]. While MPRA in the STARR-seq style has been widely adopted, it is important to consider that mRNA sequences could be affected by posttranscriptional effects such as mRNA degradation which would be indistinguishable from transcriptional effects [57].
Silencer MPRA has been applied to detect thousands of CREs acting as silencers, which were enriched for disease-associated SNPs [42], highlighting the need to decipher regulatory logic of transcriptional silencing in understanding disease etiology [42].
MPRA for studying posttranscriptional regulationSplicingMPRA can be combined with methods that sequence populations of cells binned by fluorophore expression, called Sort-seq [58], to study posttranscriptional processes like alternative splicing. In splicing MPRA, a red fluorophore (mCherry) is constitutively expressed, and a three-exon, two-intron minigene construct is cloned into a plasmid in such a way that when the middle (tested) exon is skipped, a green fluorophore (GFP) is also expressed (Fig. 3E) [43]. Variants can be located in the variable intron sections on either side of the test exon or within the exon. Cells are sorted into bins using GFP:mCherry ratios by fluorescence-activated cell sorting (FACS), where a higher ratio indicates greater intron excision. Plasmid DNA is then sequenced within each bin to determine which variants affect splicing. In an experiment utilizing this assay, many of the variants that lead to differences in splicing were located outside of canonical splice sites in both exons and introns, demonstrating that novel types of genetic variation affect splicing [43].
Though splicing MPRA have not yet been used to validate neurodevelopmental disorder-associated variant function, alternative splicing is a critical process for neuronal fate specification during neurogenesis [59, 60], and differences in alternative splicing have been identified in postmortem brains from individuals with autism, schizophrenia, and bipolar disorder [61]. Rare neurodevelopmental disorders can also be caused by alterations in alternative splicing. For example, familial dysautonomia, a degenerative sensory and autonomic nervous system disorder, is caused by a 5′ splice site mutation in an intron of IKBKAP [62]. The mutation results in variable exclusion of exon 20 and reduced IKAP protein levels in neuronal tissue [63]. Identifying the mutation has allowed understanding of the disease mechanism [64] and testing of therapeutic treatments [
留言 (0)