
記住我


All data supporting the findings of this study are available within the paper and Data Supplement. Additional inquires can be directed to the corresponding author.
AnimalsEighty-six male Wistar rats (350–400 g) were purchased from the breeding facility at the Medical University of Silesia in Katowice, Poland. The rats were maintained in temperature- and humidity-controlled animal quarters, under standard light/dark conditions (12 h/12 h), with food and water ad libitum. The Local Ethical Committee on the Care and Use of Animals for the Medical University of Silesia in Katowice, Poland approved all procedures performed during the study. All animal procedures were performed at Department of Experimental Medicine of Medical University of Silesia in Katowice, Poland.
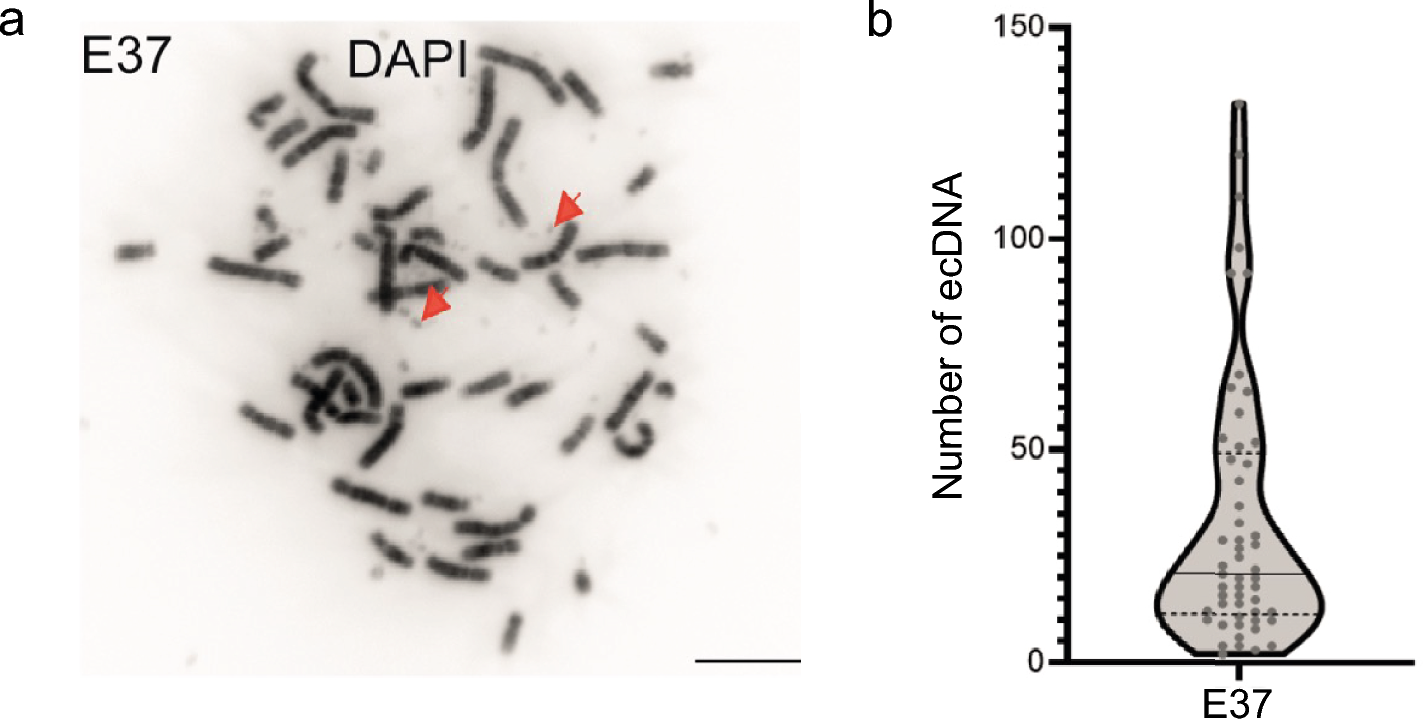
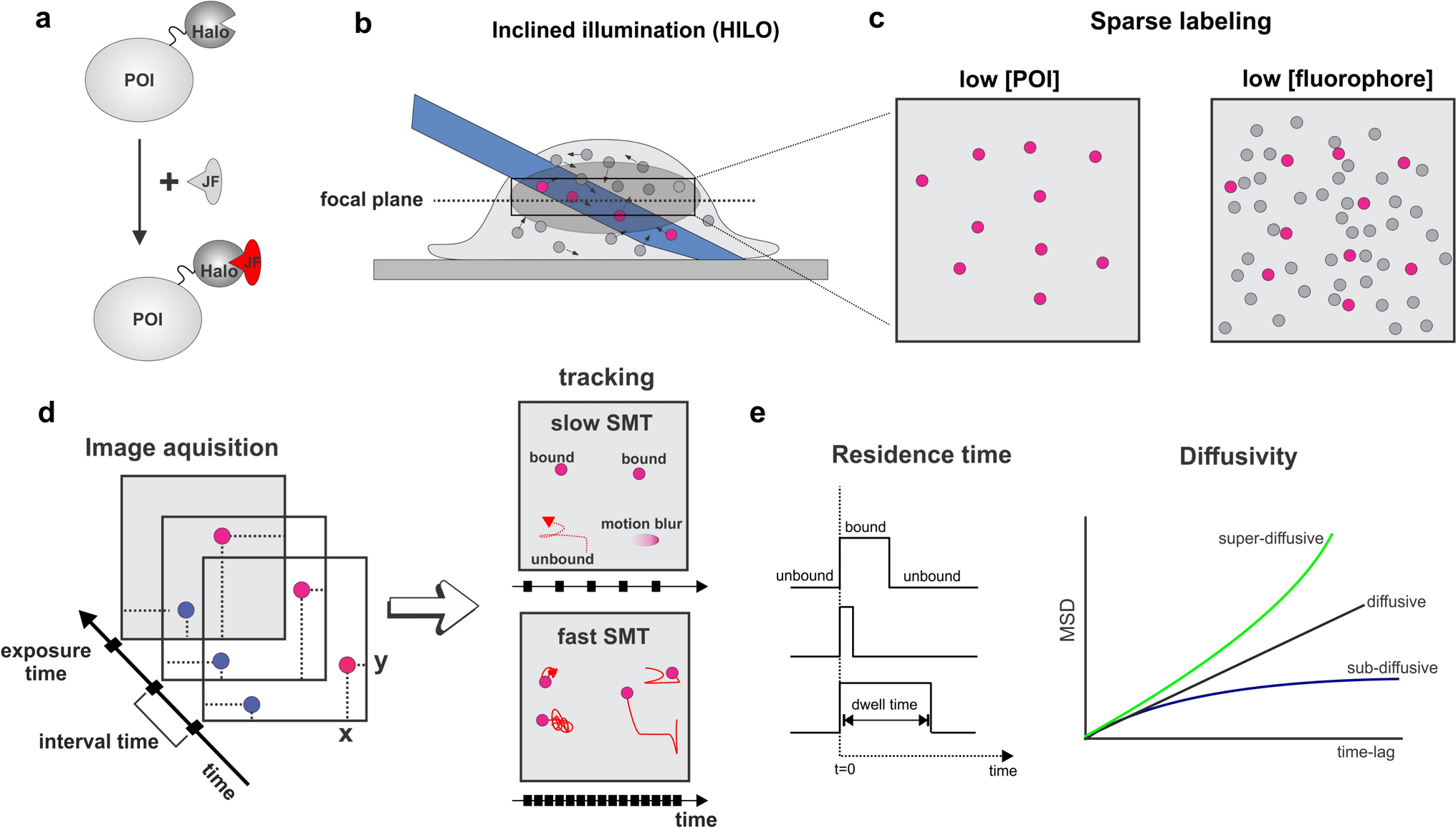
Prechiasmatic cistern SAH modelEighty-six rats were included in the study. Before surgery, rats received an intraperitoneal injection of a mixture of ketamine (60 mg/kg) and xylazine (10 mg/kg). Then, the rats were placed in a stereotaxic frame, in a prone position. A small incision was made on the top of the skull to expose the midline and bregma. Using a stereomicroscope, a small hole (2 mm diameter) was made, using a dental drill, 0 mm from the midline and 5 mm anterior from bregma. A 0.6-mm-diameter, sterile, heparinized, polyvinyl cannula was inserted into the prechiasmatic cistern, as previously described (Prunell et al. 2002). The tube was tilted 45° in the sagittal plane, placed in the hole, and lowered until the tip of the tube reached the base of the skull, 2 mm anterior to the chiasm. The free outflow of cerebrospinal fluid through the installed catheter verified the correct position of the cannula. Before placing the animal into the supine position, bone wax was applied around the hole in the skull, to avoid the uncontrolled movement of the cannula. Arterial blood (300 µL) was collected from a femoral artery of an animal subjected to the procedure. Directly after blood withdrawal, 250 µL of nonheparinized arterial blood was slowly injected into the prechiasmatic cistern over a 20-s period, under aseptic technique. Then, the cannula was removed, the hole was filled with bone wax, and the skin of the skull was sutured with a surgical thread. Control animals (SHAM) received an injection of 250 µL saline into the prechiasmatic cistern, instead of blood. The animals were subcutaneously injected with 10 mL 0.9% NaCl following the operation, to prevent dehydration, and then returned to their cages. Mortality rate after SAH was 14%. After 24 h from ictus, rats were sacrificed under deep anesthesia and brains were removed. High-resolution pictures of the base of the brains depicting the circle of Willis and basilar arteries were taken to evaluate the extent of SAH using a grading system as previously described (Sugawara et al. 2008; Egashira et al. 2015). The basal brain including brainstem was divided into six segments. Each segment was assigned a grade from 0 to 3, depending on the amount of blood. The minimum SAH grade is 0, and maximum grade is 18. In the present study, only rats that were graded 8 or more were included in the study. We did not observe statistically significant difference between rats from SAH and SAH + minocycline (Mino) groups (data not published). The hemorrhagic blood covered the inferior basal temporal lobe, and brain tissue adjacent to clotted blood was used for later morphological and biochemical analyses (Wang et al. 2012) (Fig. 1).
Fig. 1
SAH model in rats. a, b A schematic representation of the areas taken for assay, SAH or SAH + Mino and control rats. c Hematoxylin and eosin staining of SAH brain. Black arrow points to hemorrhagic blood present within subarachnoid space of brain. d Electron micrograph represents the subarachnoid space filled with erythrocytes (black arrows). Scale bar, 5 μm (d)
Minocycline administrationRats that underwent SAH induction were randomly divided into SAH or SAH treatment group. Rats from the treatment group received minocycline (Sigma-Aldrich, USA), at a dose of 1 mg/kg, in 1 mL 0.9% NaCl, injected intraperitoneally, 10 min after SAH induction. The animals from the vehicle group, SHAM, received intraperitoneally the same volume of saline 10 min after SAH induction.
Pain managementParacetamol in oral suspension was used as a pain relief agent for rats included in the project. One milliliter of paracetamol (100 mg/ml) was added to the 250 ml of drinking water, which was provided ad libitum.
Transmission electron microscopyFor transmission electron microscopy (TEM) studies, animals (n = 3–4 per group) were transcardially perfused, under constant pressure 130 mmHg for 10 min, with 100 mL ice-cold phosphate-buffered saline (PBS), pH 7.4, followed by 200 mL 4% paraformaldehyde solution, pH 7.4. Immediately after brain removal, brain was sliced into 1.5-mm-thick slices and post-fixed in 2.5% glutaraldehyde (SERVA Electrophoresis, Germany) in cacodylate buffer, pH 7.4, for 4 h, at room temperature and then washed several times in the same buffer. Then, tissue samples from cortex within the basal temporal lobe and adjacent to clotted blood from each hemisphere were cut into smaller 1–1.5 mm3 blocks (8–12 blocks per animal). Subsequently, the tissue was fixed in 1% osmium tetroxide (Polysciences Inc., USA) and dehydrated in a graded ethanol series (50%, 70%, 90%, 96%, and 100%) and propylene oxide. Propylene oxide to Epon 812 mixtures, at 2:1 (v:v) and 1:2 (v:v), was applied to infiltrate the samples, which were later embedded in Epon 812 epoxy resin (SERVA Electrophoresis, Germany), and polymerized for 48 h at 60 °C. Ultrathin sections were cut from representative samples, using a diamond knife (45°; RMC, USA) on a Reichert OmU-3 ultramicrotome (Reichert Austria). Sections were mounted on 300-mesh copper grids and stained with 0.5% aqueous uranyl acetate and lead citrate (LAURYLAB Saint, France), using a Leica EM AC 20 Stainer (Leica Microsystems, Austria). The grids were air-dried and later examined with a TECNAI G2 12 Spirit BioTWIN transmission electron microscope (FEI, the Netherlands), at 120 kV. The Morada CCD camera (Olympus Soft Imaging System Solutions GmbH, Germany) was used to capture images (tif format, 8-bit, 2656 × 3360 pixels, with 1.5 s integration time, camera binning 1) from representative regions using TEM Imaging and Analysis software (TIA software, version 3.1) (FEI, the Netherlands) in magnification range from 16,500× to 43,000× (Sawczyn et al. 2018).
Sampling procedureA researcher blinded to condition chose three to five ultrathin sections from the single mesh grid and photographed capillaries. To avoid bias of double measurements of a single capillary, some sections were discarded between those collected and placed on mesh grid. The researcher used a software tracking tool to record and control move within sections on the grid. The researcher photographed tissue samples within the basal temporal lobe and adjacent to clotted blood for later analysis. A capillary lumen met the inclusion criteria when it was lined with no more than two endothelial cells, surrounded by a basement membrane, cut in cross-section, and did not contain blood cells or plasma. Included samples were photographed under 6000× magnification, to obtain a general view of the capillary and surrounding neuropil, under 11,500× magnification for quantitative analysis, and under 43,000× magnification to identify specific structural traits. Images with an endothelial or pericyte nucleus were excluded from the quantitative analysis because they generated extreme measurements (e.g., for endothelial or pericyte area/coverage/length adhesion) and were too infrequent to adequately sample (Nahirney et al. 2016).
Morphometric analysisGiven the lack of quantitative analysis that has been performed for capillary ultrastructure in the brain after SAH, we applied a methodological approach of sampling and morphometric analysis that was previously employed to study ultrastructural changes in the brain after ischemia. For quantitative analysis, the ultrastructural traits of the capillary and perivascular area were thresholded and measured using ImageJ software, by an experimenter blinded to condition (Schneider et al. 2012). Other qualitative ultrastructural traits that were observed in the electron micrographs following SAH induction but were difficult to measure were described and compared with capillary profiles in the control (SHAM) and minocycline groups (SAH + Mino).
The ImageJ software functions were used to measure the circularity and roundness of the abluminal and luminal endothelium membranes (capillary and lumen, respectively), where circularity \(\left(4\pi \frac}}^}\right)\) ranged from 0 (infinitely elongated polygon) to 1 (perfect circle) (Haley and Lawrence 2017), and roundness \(\left(4\frac}\right)}^}\right)\) was the inverse aspect ratio of the particle-fitted ellipse (Schneider et al. 2012). Then, the capillary and lumen areas were manually outlined in every electron micrograph, to calculate the ratio of endothelium volume fraction \(\left(\frac}}\right)\) and lumen volume fraction \(\left(\frac}}\right);\); hence, the endothelium area was obtained by subtracting the lumen area from the capillary area. These parameters were used to determine changes in the volumes occupied by the endothelium or lumen within the studied capillaries. The capillary area/circumference and lumen area/circumference data were used to calculate the ratios of lumen size \(\left(\frac}}\right)\) and capillary size \(\left(\frac}}\right)\), to account for variations in the capillary and lumen sizes. The grid overlay image analysis (GOIA) technique was applied to measure the basal lamina (BL) thickness (Baum and Bigler 2016). A 0.5 µm × 0.5 µm grid was placed on to capillary electron micrograph, and a minimum of three spots where the gridline crossed the BL was used to calculate the average value of the BL thickness. Places, where the grid crossed the BL adjacent to a pericyte were excluded from analysis because this location is much thinner (Frank et al. 1987; Alba et al. 2004). TJ complexity (TJ tortuosity) was measured, as previously described (Jackman et al. 2013), where TJ tortuosity was calculated by dividing TJ length by the diagonal of the rectangle that contains the complete TJ, from where it starts at the luminal side to where it ends at BL. Pinocytic vesicle density was defined as the accumulated number of vesicles per square micrometer, counted in three to five selected fields of the endothelial cytoplasm of one capillary and added together to obtain the reference area (Alba et al. 2004). The structure under study was outlined to measure the pericyte processes area, and when more than one pericyte was present on the profile of a capillary, their areas were added together (Frank et al. 1987). The pericyte processes area was divided by the capillary area to determine the pericyte volume fraction, which represents the volume occupied by pericyte processes inside the capillary wall. The ratio of pericyte or astrocyte coverage was quantified by tracing the pericyte length adhesion or astrocyte length adhesion, including processes, in contact with the endothelial BL and dividing the sum length by the circumference of the BL (Alba et al. 2004; Nahirney et al. 2016). In addition to pericytes, the perivascular space contains astrocyte endfeet. In the majority of electron micrographs from the SAH and SAH + Mino groups, the astrocyte endfoot area was impossible to define because the space adjacent to the capillary was deteriorated; therefore, we used the ImageJ threshold function to measure unequivocally bright spaces adjacent to capillary (the electron-lucent area), which were later called “Holes.” Means from multiple regions of interest (ROI) from one tissue section were used to calculate mean value for each parameter per one animal, used for later analysis.
ImmunofluorescenceAnimals (n = 3–4 per group) were transcardially perfused with 100 mL ice-cold PBS, pH 7.4, followed by 200 mL 4% paraformaldehyde solution (Merck, Sigma-Aldrich, Germany), and 100 mL 4% paraformaldehyde, with 10% sucrose (Sigma-Aldrich). Brains were quickly removed and post-fixed in 4% paraformaldehyde, with 10% sucrose, overnight at 4 °C. Subsequently, brains were washed in PBS, pH 7.4, and later dehydrated in a graded sucrose series (10%, 20%, and 30%), in PBS, pH 7.4 (weight/volume), until complete dehydration was achieved. Fixed and cryoprotected brains were embedded in TissueTek (Sakura, California, USA) and coronally cryosectioned into 30-µm-thick slices. Collected sections were washed in PBS, pH 7.4, and preserved in PBS, pH 7.4, with 0.05% sodium azide, at 4 °C, for further triple immunostaining procedures on free-floating tissue sections. The staining procedure was performed on an orbital shaker. Briefly, washed sections were pretreated with PBS, pH 7.4, containing 0.1% Triton X-100 (Sigma-Aldrich) for 2 h and then blocked in 10% serum, with 1% Triton X-100, for 1 h. Next, sections were washed in PBS, pH 7.4, and incubated at 4 °C overnight with primary antibodies diluted in PBS, pH 7.4, containing 1% serum and 0.1% Triton X-100. The following antibodies were used (manufacturers provided proof of validation on the technical specification insert): EMMPRIN (1:50, mouse, Bio-Rad, USA), EMMPRIN (1:100, rabbit, Santa-Cruz, USA), collagen IV (1:50, donkey, Bio-Rad), occludin (1:100, rabbit, Antibodies-online, Germany), Pan-Laminin (1:200, rabbit, Sigma-Aldrich), claudin-5 (1:100, rabbit, Antibodies-online), MMP-9 (1:200, rabbit, Abcam, UK), MMP-2 (1:200, rabbit, Abcam), GFAP (1:200, rabbit, Abcam), MAP-2 (1:200, mouse, Invitrogen, USA), AIF-1/Iba-1 (1:200, Antibodies Online), and CD-45 (Novius Biologicals, USA). Antibodies were carefully chosen to avoid cross-reactivity in triple immunofluorescence staining. After washing with PBS, pH 7.4, containing 0.1% Triton X-100 (4 × 5 min), sections were incubated for 1 h with three different secondary antibodies (conjugated with fluorochrome) simultaneously: DyLight conjugated with goat anti-donkey IgG (1:1,000, Jackson Immunoresearch, UK), Alexa 488 conjugated with goat anti-rabbit IgG (1:1,000, Jackson Immunoresearch), and tetramethylrhodamine conjugated with goat anti-mouse IgG (TRITC, 1:1,000, Jackson Immunoresearch). Gentle orbital shaking was used during fixation and staining procedures. After staining, sections were mounted on a microscope slide, after careful washing with PBS, pH 7.4, containing 0.1% Triton-X 100, and PBS, pH 7.4, covered with Vectashield Mounting Medium (Vector Laboratories, USA), and closed with coverslips. Potential unspecific binding of secondary antibodies was evaluated by omission of the primary antibody in sections that were otherwise processed similarly. Sections were stored at 4 °C, in the dark, until further viewing on a confocal microscope.
Confocal microscopy image acquisitionThe sections were photographed under Olympus Fluoview FV1000 confocal microscope, (Olympus Europa Holding, Germany), equipped with Olympus FV1000 SIM scanner system (Olympus Europa Holding) using FV10-ASW 4.2a acquisition software (Olympus Europa Holding), with the following argon lasers: 405 nm, 488 nm, and 559 nm and objective lens: magnification 30; NA 1.05; super apochromat; model UPLSAPO3XS. Additional image acquisition information: PMT voltage 140–160; voxel size 0.414 µm XY, 0.82 µM Z; resolution 1024 × 1024; image bit depth 12 bit. Three different regions within the basal temporal lobe that were adjacent to clotted blood were scanned every 0.2 µm in the z-axis, on multiple focal planes, and in three channels simultaneously, to perform three-dimensional (3D) colocalization analysis. Both the immunostaining procedures and image acquisition with confocal microscopy were performed using identical settings for all samples. Therefore, relative comparisons between different samples treated with the same antibody could be performed. Subsequently, confocal z-stacks underwent the denoise procedure and the 3D deconvolution procedure in Autoquant X3.1 (Media Cybernetics Inc, USA) and were analyzed in Imaris 9.3 (Bitplane AG, Switzerland). The extent of colocalization between two labels (DyLight and Alexa 488 or TRITC) was measured using a threshold-based colocalization algorithm, as previously described (Raji et al. 2008). The results were presented as colocalization percentages and Pearson’s correlation coefficients (r). r values equal to or greater than 0.5 were considered as two fluorescent signals correlating between each other, varying in the same voxel. Mean values for percentage of material colocalized and Pearson’s correlation coefficient (r) are included in Supplementary Table I and every scatterplot of Supplementary Figs. I–III. The extent of colocalization of two stains was assessed using 3D colocalization and spot detection, implemented in Imaris software, which allowed us to analyze the percentage of material colocalized in whole stacks of confocal sections, as described by Costes et al. (2004).
ImmunoblottingThe animals were transcardially perfused (n = 10–16 per group) with 100 mL PBS, pH 7.4, and brain samples were cut and stored for later protein and mRNA expression analyses. Then, 50–60 mg of frozen brain sample was preincubated in the ice-cold lysis buffer for 15 min, pH 7.4 (20 µL/1 mg of tissue), consisting of 20 mM Tris–HCl, 150 mM NaCl, 2 mM 1% Triton X-100, 10% glycerol, 1 mM PMSF, Protease Inhibitor Cocktail, and PhosSTOP (Sigma-Aldrich), in quantities recommended by the manufacturer, then mechanically homogenized and incubated for additional 20 min in ice-cold lysis buffer. Tissue lysates were centrifuged at 12,000g, at 4 °C, for 10 min. The protein concentration was estimated by the Bradford method, using the Protein Assay Kit (Bio-Rad). The samples containing 30 µg per lane were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) (Carl Roth, Germany) and electrotransferred onto a polyvinylidene difluoride Immobilon-FL Transfer Membrane (Merck, Millipore Solutions, Germany). The membranes were rinsed in PBS (pH 7.4) and blocked in PBS Odyssey Blocking Buffer (pH 7.4) (LI-COR Biosciences, USA), for 1 h at room temperature. Next, the membrane was incubated at 4 °C for 24 h with the following primary antibodies diluted in PBS, pH 7.4 Odyssey Blocking Buffer, with 0.02% Tween 20: EMMPRIN (1:500, rabbit, Santa Cruz), occludin (1:2000, rabbit, Antibodies-online), and claudin-5 (1:2000, rabbit, Antibodies-online) (Sigma-Aldrich). Membranes were washed in PBS, pH 7.4, containing 0.01% Tween 20 (4 × 5 min), and then incubated for 1 h with secondary antibody conjugated to IRDye 800 CW (1:15,000, goat, rabbit, LI-COR), diluted in PBS, pH 7.4 Odyssey Blocking Buffer containing 0.02% Tween 20 and 0.01% Triton-X 100. After washing in PBS, pH 7.4, containing 0.01% Tween 20 (4 × 5 min), and then PBS, pH 7.4 (2 × 5 min), the membranes were dried and scanned with the Odyssey CLx Imaging System (LI-COR). Scanned membranes were activated once more in methanol and then incubated in stripping buffer, containing 0.1 M glycine, pH 2.9, for 1 h at room temperature. Immunoblotting for β-actin (1:2,000, rabbit, Cell Signaling, USA), as a loading control, was performed as described above. ImageStudioLite (LI-COR) was used to quantify the fluorescence intensities of each band, adjusted by the membrane background value measured above and beneath each sample, and normalized against the intensity of the loading control. The average values of protein levels in SHAM control samples, on the same membrane as the SAH and SAH + Mino samples, were used to calculate the fold changes in protein levels.
Quantitative real-time PCRFollowing transcardial perfusion, n = 10–16 per group, 15–20 mg of the cortex was rapidly frozen on dry ice and protected in RNA later (EURx, Polska) buffer, until the mRNA isolation protocol was performed. mRNA was extracted using the Total RNA Mini kit (A&A Biotechnology, Poland), according to the manufacturer’s recommendations. Reverse transcription (of 100 ng/µL) was performed with the NG dART RT Kit (EURx), in a DNAEngine DNA thermocycler (Bio-Rad). Quantitative real-time PCR was performed to measure basigin 2 (BSG2), occludin, claudin-5, MMP-2, and MMP-9 transcriptional levels using the CFX96TM Real-Time System (Bio-Rad), in default mode, with SYBR Green Master Mix, using following primers (Genomed, Poland): GAPDH, forward 5′-TGGAAAGCTGTGGCGTGAT-3′ and reverse 5′-AACGGATACATTGGGGGTAG-3′; OCLN, forward 5′- TAGCCATTGTCCTGGGGTTCAT-3′ and reverse TTTCTTCGGGTTTTCACAGCAAA-3′; CLDN5, forward 5′TAAGGCACGGGTGGCACTCA-3′ and reverse 5′-CTACGATGTTGGCGAACCAG3′; BSG2, forward 5′-GTTTGTGAAGCTGATCTGCAAG3′ and reverse 5′-ACAGCTCAGGCGTGGATATAAT-3′; MMP-2, forward 3′-GCAACCACAACCAACTACGA-3′ and reverse 5′-CCAGTGTCAGTATCAGCATCAG-3′; MMP-9, forward 5′-GCAAACCCTGCGTATTTCCAT-3′ and reverse 5′-CCATCCGAGCGACCTTTAGTG-3′. mRNA levels were measured relative to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA levels, which served as an internal control. Relative mRNA levels were calculated using the comparative CT method and expressed using the 2−ΔΔCT method, as fold changes relative to control samples (Livak and Schmittgen 2001).
Gelatin zymographyProtein extraction was performed, as described previously (Wiera et al. 2013), with some modifications. Tissue homogenization was performed in ice-cold 50 mM Tris–Cl buffer, containing 10 mM CaCl2, 0.25% Triton X-100, and 0.1 mM PMSF (30 µL buffer per 1 mg wet tissue). Homogenates were centrifuged at 6000g, for 30 min at 4 °C, and the pellet containing the Triton X-100-insoluble proteins was then resuspended in 50 mM Tris–Cl buffer, pH 7.4, containing 2% Triton X-100 and 0.1 M CaCl2, incubated for 12 min at 60 °C, and centrifuged (30 min, 10,000g, 4 °C). The Amicon Ultra-0.5 3 K (Millipore Sigma) column was used to concentrate the supernatant, containing the extracellular matrix and membrane-associated proteins. Protein concentrations were measured using the Protein Assay Kit (Bio-Rad). Equal amounts of proteins were separated, under nonreducing conditions, using an 8% polyacrylamide (Carl Roth) gel that was copolymerized with 2% fish-skin gelatin (Sigma-Aldrich). After electrophoresis, gels were washed in 2.5% Triton X-100 (2 × 30 min) and incubated in enzymatic buffer (50 mM Tris, pH 7.5, 10 mM CaCl2, 1 µM ZnCl2, 1% Triton X-100, and 0.02% sodium azide), at 37 °C, for 3–5 days, with continuous, gentle mixing in the STUART Orbital Incubator SI50 (Cole-Palmer, UK). The gels were first stained with 0.1% Coomassie Blue G-250 (Carl Roth) for 2 h and then destained with 5% acetic acid, until white proteolytic bands appeared on the blue background, allowing the visualization of the active MMP levels. Additionally, the same samples were resolved in a parallel SDS–PAGE gel (Carl Roth), without gelatin, to verify protein loads across samples. The Geldoc1000 system (Bio-Rad) was used to digitally photograph gels, and ImageJ (Schneider et al. 2012) was used to quantify the gelatinolytic activity.
Statistical analysisStatistical comparisons were conducted with analysis of variance (ANOVA) test, GraphPad Prism 9. For each comparison between groups, a false discovery rate (FDR) correction was performed. All statistical tests were calculated on the basis of means generated from the number of animals in each group. Differences at P < 0.05 were considered to be statistically significant.
留言 (0)