
記住我










Familial adenomatous polyposis (FAP) is an autosomal dominant cancer predisposition syndrome caused by pathogenic variants in the APC gene. FAP patients with a classical phenotype present with hundreds to thousands of colorectal adenomatous polyps at a mean age of onset of 16 years. Depending on the position of the causative variant in the APC gene, patients can also have an attenuated phenotype with lower polyp burden and later age of onset. As colorectal adenomas are potential precancerous lesions, untreated patients have a high risk for colorectal cancer (CRC). Therefore, total colectomy is recommended if polyposis can no longer be controlled endoscopically. FAP patients can also develop adenomas in the upper gastrointestinal tract, especially the duodenum, in addition to desmoid tumours that frequently occur following surgical procedures. Further symptoms associated with FAP include congenital hyperplasia of the retinal pigment epithelium, jaw osteomas and gastric fundic gland polyps.
Approximately 20% of patients with clinical FAP remain unsolved after routine molecular genetic analysis of the APC and additional polyposis genes, suggesting additional loss-of-function mechanisms.1 Aside from an abundance of coding variants, several intronic rearrangements, a complex event that generates a complete deletion of exon 5 (c.422+1123_532–577 del ins 423–1933_423–1687 inv), or deep intronic single nucleotide variants have been described for APC.2–6 The latter loss-of-function mechanisms may frequently escape detection in routine diagnostics.
Deletions differing in size but all comprising non-coding exon 1B of APC and the exon 1B-associated promoter region were reported in numerous FAP families.7–15 mRNA expression analysis of various tissues of patients from these families strongly suggested allelic silencing of APC mRNA expression due to deletion of the promoter/exon 1B region as another pathomechanism in FAP patients. Similar reductions in allele-specific APC expression were observed due to pathogenic single nucleotide variants in APC promoter 1B in patients with FAP16 or gastric adenocarcinoma and proximal polyposis of the stomach.17 18
In the present study, we summoned an extensive molecular diagnostics tool suite to solve a case of FAP that had remained molecularly undiagnosed by routine genetic testing. We found a complex genomic rearrangement involving the fragmented insertion of a ~3.75 Mb region of chromosome 5 into chromosome 10. The rearrangement, which meets all criteria of chromothripsis,19 disrupts the APC gene thereby dissociating promoter/exon 1B from the coding region of the gene. We propose that the resulting allele-specific reduction in APC mRNA expression is a major contributing factor to the patient’s FAP.
MethodsPatient samplesGenetic counselling and consecutive genetic diagnostic and predictive testing of the index patient and family members was performed with consent according to German laws. Sulindac was administered as an off-label use within the framework of an observational study approved by the ethics committee.
Short-term culture of peripheral blood mononuclear cells (PBMCs)PBMC cultures were prepared by separating 3 mL heparinised whole blood in Leucosep tubes followed by isolation of the intermediate cell layer and one wash in sterile saline. Washed cell pellets were transferred into 5 mL PB-MAX media (ThermoFisher) in a 15 mL conical tube and incubated at 37°C for 72–96 hours. Parallel cultures received 0.2 mg/mL puromycin for 5 hours prior to harvesting to inhibit nonsense-mediated mRNA decay. Cells were pelleted by centrifugation and frozen at −20°C until RNA extraction.
Chromosomal microarray analysis (CMA)CMA was performed using Infinium CytoSNP-850K BeadChip v1.2 (Illumina) according to the manufacturer’s instructions. Scanning and image acquisition was done on an Illumina iScan microarray scanner. Data analysis was performed using BlueFuse Multi software V.4.5 (Illumina).
Fluorescence in situ hybridisationFISH analysis of the APC gene was performed with BAC clone RP11-3B10 (Empire Genomics) labelled in Orange-dUTP. The probe was hybridised to metaphase chromosomes of peripheral blood lymphocytes according to the manufacturer’s instructions. Slides were visualised under an epifluorescence microscope (Zeiss Imaging2). Images were captured and analysed with PathVision software (Applied Imaging).
Ultra-high molecular weight DNA extraction and labelling for optical genome mappingFor each sample, a minimum of 650 µL of whole peripheral EDTA blood was used to purify ultra-high molecular weight DNA using the SP Blood & Cell Culture DNA Isolation Kit (Bionano Genomics). Briefly, after counting, white blood cells were pelleted (2200 g for 2 min) and treated with LBB lysis buffer and proteinase K to release genomic DNA (gDNA). After inactivation of proteinase K by PMSF treatment, genomic DNA was bound to a paramagnetic disk, washed and eluted. Ultra-high molecular weight DNA was left to homogenise at room temperature overnight. The next day, DNA molecules were labelled using the DLS (Direct Label and Stain) DNA Labeling Kit (Bionano Genomics). Seven hundred fifty nanograms of gDNA were labelled in the presence of direct label enzyme (DLE-1) and DL-green fluorophores. After clean-up of excess DL-green fluorophores and rapid digestion of the remaining DLE-1 enzyme by proteinase K, the DNA backbone was counterstained overnight before quantification and visualisation on a Saphyr instrument. A volume of 8.5 µL of labelled gDNA solution of concentration between 4 and 12 ng/µL was loaded on a Saphyr chip and scanned on the Saphyr instrument (Bionano Genomics).
De novo assembly and structural variant callingThe de novo assembly was executed on Bionano Solve software V.3.6. Reporting and direct visualisation of structural variants was done on Bionano Access V.1.6. The following filter settings were used: insertion/deletion=0, inversion=0.01, duplications=−1, translocation=0, CNV=0.99.
Long-read genome sequencing (GS)Genomic DNA was purified from peripheral blood samples using the NucleoMag Blood 3 mL Kit (Macherey-Nagel), and 1 µg gDNA was subjected to sequencing. DNA end-prep was performed using the NEBNext Ultra II End Repair/dA-Tailing Module (E7546, New England Biolabs) followed by adapter ligation with the NEBNext Quick Ligation Module (E6056, New England Biolabs). Sequencing libraries were prepared with the Ligation Sequencing Kit 1D (SQK-LSK109, Oxford Nanopore Technologies) and run on R9 flow cells on the Oxford Nanopore Technology GridION instrument. DNA purifications at each step of library preparation were performed using Agencourt AMPure XP beads (A63882, Beckman Coulter). Sequencing was performed to a final mean sequencing depth of 7.13 x. Structural variant (SV) calling was performed with Nanovar V.1.3.820 using Minimap2 (2.17-r941),21 blast+ (2.10.1)22 and hs-blastn (v0.0.5).23 The human reference sequence was GRCh38. The final structure and exact breakpoint information was deduced from a combination of Bionano and Nanovar information, manual inspection of soft clipped read sequences at predicted breakpoints using the Integrative Genomics Viewer V.2.9.024 and inspection of Sanger sequences of PCR fragments spanning the breakpoints (see further).
PCR confirmation of genomic breakpointsGenomic breakpoints identified by GS were confirmed by PCR amplification and DNA sequencing. Amplicons were generated using the primers listed in online supplemental table S1 with Taq DNA polymerase (Qiagen). After denaturation at 94°C for 3 min, 35 cycles were performed (94°C for 45 s, 56°C for 45 s, 72°C for 1 min 30 s) including a final extension at 72°C for 10 min. After digesting the PCR Product with exonuclease, 1 µL of the amplified product was sequenced using the BigDye Terminator V.1.1 Cycle Sequencing Kit (Applied Biosystems) according to the manufacturer’s instructions. Upstream and downstream PCR primers were used for sequencing except for the fragment spanning BP1 (chr5: I junction) for which a separate sequencing primer was designed (denoted by an asterisk in online supplemental table S1).
RNA sequencingRNA extraction from PBMCs was performed following the instructions of the RNeasy RNA blood mini kit (QIAGEN). RNA from PAXgene blood tubes was extracted using the PAXgene Blood RNA System (Qiagen) following the manufacturer’s protocol. RNA abundance was measured photometrically (Xpose), and RNA integrity number (RIN) was determined (Agilent Bioanalyzer).
Cancer-related transcripts were enriched and sequenced according to the method described in detail in ref 25. Briefly, 50 ng total RNA were reversely transcribed with oligo-dT using kit SQK-PCB109 from Oxford Nanopore Technologies and amplified by PCR. cDNAs were captured with a custom Agilent SureSelect probe set directed against 123 hereditary cancer genes. Following a second PCR amplification of captured cDNAs, sequencing libraries were prepared with the SQK-PCB109 kit and sequenced on R9 flow cells on the GridION. Capture and sequencing was performed in triplicates. Differential expression analysis has been performed against 21 reference samples in triplicates using the Oxford Nanopore Technologies Pipeline for differential gene expression (DGE) and differential transcript usage analysis of long reads (https://github.com/nanoporetech/pipeline-transcriptome-de), which uses minimap2 V.2.18,21 salmon V.1.5.026 and edgeR V.3.34.0.27
RT-PCR analysisRT-PCR analysis from the patients’ cDNA was performed as described previously28 with minor adaptations to the APC transcript. From 1 µg of total RNA, cDNA was generated using the iScript Select cDNA synthesis kit (Bio-Rad) and an Oligo-(dT) primer.29 To amplify the APC transcript fragments of interest, primers in exon 9 and 14 were used to obtain a c.1189_1943 amplicon with Ampli-Taq Gold polymerase (ThermoFisher) in a Touchdown-PCR from 64°C to 54°C.30 PCR products were visualised on a 1% agarose gel, purified with Exo-SAP (USB), and sequenced with Big Dye V.1.1 (Applied Biosystems). Sequences were run on an ABI PRISM 3100 Avant or ABI 3730. Sequences were inspected visually with Sequence Scanner V.2.0 (Applied Biosystems), and the allelic balance of known heterozygous variants of exon 11 and 13 was estimated.
ResultsCase descriptionA female patient, 30 years old at diagnosis, presented with an adenoma burden resembling an attenuated FAP. Multiple polyps (~100) were found mainly in the proximal colon with few also in the duodenum in addition to numerous gastric hyperplastic gland polyps. The patient was treated with sulindac as an off-label use for 15 years. Polyp burden in the colon decreased significantly under this therapy allowing for removal of polyps in yearly colonoscopies. Due to the increasing number of duodenal adenomas, pancreas sparing duodenectomy was carried out at the age of 50 years. Four years later, the patient developed intestinal gastric cancer with neuroendocrine parts (mixed neuroendocrine non-neuroendocrine neoplasm). Sanger sequencing of the APC gene at the time of initial diagnosis revealed no pathogenic variants. Years later, next-generation sequencing (NGS) panel analysis comprising APC and other polyposis-associated genes again failed to detect any pathogenic variants. Family history was conspicuous for hereditary tumour predisposition. The patient’s mother was diagnosed with CRC in her mid-40s; clinical files did not document FAP. The father died of CRC at an age between 70 and 75 years. The patient has healthy children (currently between 15 and 30 years old); none of them developed colorectal polyposis so far.
Genomic analysis defines a complex chromosomal rearrangement disrupting the APC locusSince NGS of polyposis genes did not reveal any pathogenic germline variants in the patient, we assessed chromosomal copy number variants by microarray analysis. This revealed a 105 kb deletion in chromosome band 5q22.1 (genomic position hg38 chr5: 111,651,620–111,758,049; online supplemental figure S1) encompassing exons 1–4 of the NREP gene (NM_004772.4; OMIM 607332) as well as a large part of the STARD4-AS1 lncRNA transcript both of which are most highly expressed in the cerebellum (https://www.gtexportal.org/home/gene/ENSG00000246859, https://www.proteinatlas.org/ENSG00000134986-NREP/summary/rna). Since neither gene has been implicated in hereditary colon cancer, their contribution to the patient’s phenotype was deemed unlikely. In addition, the distal breakpoint of the NREP/STARD4-AS1 deletion maps 953 kb upstream of the APC gene thus rendering a causal involvement in polyposis implausible.
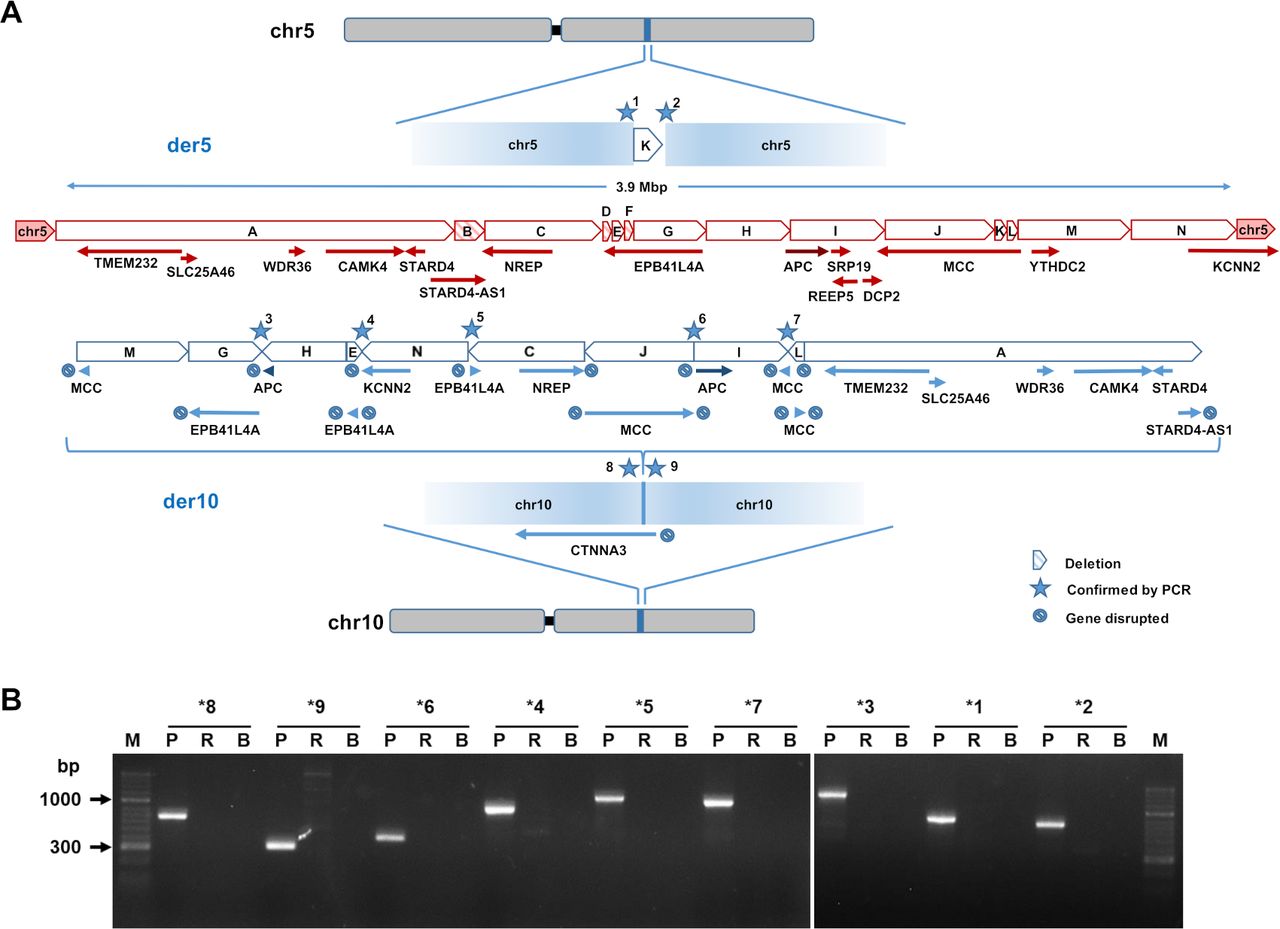 Figure 1
Figure 1 Structure of the chromosomal rearrangement. (A) The drawing in red depicts the normal structure of chromosome 5 (ie, the intact allele), indicating genes and the genomic fragments involved in the rearrangement. Blue elements show the derivative chromosomes 5 and 10 reconstructed from optical mapping and long-read DNA sequencing. The orientation of the fused fragments is indicated as well as genes that are disrupted by the rearrangement. Deleted fragments B, D and F are highlighted by shading. The HGVS description of the rearrangement is as follows: seq[GRCh38] del(5)(q21.1)der(10)ins(10;5)(q21.3;q22.1q22.3); NC_000010.11:g.67598332_67598333ins[NC_000005.10:g.113429662_113851075; NC_000005.10:g.112169858_112428316; NC_000005.10:g.112428345_112714041inv; NC_000005.10:g.112158505_112164089; NC_000005.10:g.113851076_114192054inv; NC_000005.10:g.111758050_112157589inv; NC_000005.10:g.113035607_113417732inv; NC_000005.10:g.112714051_113035528; NC_000005.10:g.113426215_113429671inv; NC_000005.10:g.110310618_111651619]; NC_000005.10:g.111651620_111758049del; NC_000005.10:g.112157590_112158504del; NC_000005.10:g.112164090_112169857del. (B) PCR confirmation of the breakpoints highlighted by stars in figure part A. See methods for information on primers used. B, blank; p, patient; R=reference.
Nevertheless, we reasoned that the 105 kb deletion might be an indication of a more complex structural variant on chromosome 5 that escaped detection by chromosomal microarray analysis. Turning to optical genome mapping and de novo assembly (Bionano Genomics), we found that a stretch from chromosome 5q22.1–5q22.3 (genomic position hg38 chr5: 110,310,618–114,192,054) showed structural variation. The region comprises 16 protein coding genes, in alphabetical order: APC, CAMK4, DCP2, EPB41L4A, KCNN2, MCC, NREP, REEP5, SLC25A46, SRP19, STARD4, TMEM232, TSLP, TSSK1B, WDR36 and YTHDC2. The optical map indicated that this region was fragmented into nine pieces that were deleted from chromosome 5 and reinserted into chromosome 10, band 10q21.3 (online supplemental figure S2A). The reinsertion of the fragments occurred in seemingly random order and orientation and coincided with loss of a fragment corresponding to the NREP/STARD4-AS1 deletion mapped by CMA. The insertion of chromosome 5 material into chromosome 10 was confirmed by FISH analysis (online supplemental figure S2B).
 Figure 2
Figure 2 Chromosomal breakpoint in the APC locus separating promoter 1B from the remainder of the gene body. The UCSC genome browser was used to illustrate the breakpoint observed within the APC gene (hg19). The remaining tracks show the predicted open reading frame of APC in magenta, the location of promoters 1A and 1B based on the eukaryotic promoter database43in purple and the location of H3K27ac, DNase I hypersensitive sites, POL2 and CTCF occupancy based on encode data.44 Coexistence of both APC promoters in a common topologically associating domain (TAD; bottom red) suggests similar regulatory inputs and thus promoter activity.
The Bionano map indicated that despite the dramatic rearrangement, all coding exons of the APC gene remained intact. To analyse the APC locus at higher resolution, we performed long-read GS on the Oxford Nanopore GridION platform. The sample was sequenced on three R9.4 flow cells for a total sequence of 23.2 GB (22.6 GB aligned) and an average sequencing depth of 7.13 x. Data analysis with NanoVar20 confirmed insertion of 5q22.1q22.3 material into chr10q21.3 (online supplemental figure S3). Guided by the optical map, we were able to fully assemble the rearranged loci on derivative chromosome 5 (der5) and derivative chromosome 10 (der10) at nucleotide resolution. This revealed that the entire genomic event involves 14 distinct fragments encompassing a region of ~3.9 Mb (figure 1A). The insertion into der10 harbours 10 fragments from chromosome 5 varying in length from 0.9 kb to 1.3 Mb (table 1) that were fused in apparently random order and orientation. The analysis also showed loss of fragment B within the NREP/STARD4-AS1 region as well as deletion of two small regions from the EPB41L4A gene (fragments D and F) that was not apparent in the optical map. One of the 14 fragments reinserted into der5 (fragment K, figure 1A). Nine distinct breakpoints on der5 and der10 were confirmed by PCR analysis (figure 1B). The second alleles of chr5 and chr10 retained their normal structure.
 Figure 3
Figure 3 Expression of APC mRNA. (A) Long-read RNA-seq was performed on RNA isolated from short-term cultures of the patient’s PBMCs. APC expression levels were assessed relative to 17 reference RNA samples of unaffected control subjects. Log2-fold change of APC mRNA (red dot) is shown in a volcano plot. (B) Quantification of averaged allele ratios of the SNVs in online supplemental table S2). (C) Allele ratios of APC coding SNVs c.1458T>C and c.1635G>A based on RT-PCR. Two different source materials for RNA extraction were used: PBMC short-term cultures and whole blood collected in PAXgene tubes. PBMCs, peripheral blood mononuclear cells; SNVs, single nucleotide variants.
Table 1Chromosomal fragments involved in the rearrangement
The rearrangement disrupts the structural integrity of five protein coding genes (APC, EPB41L4A, KCNN2, MCC and NREP) and one lncRNA (STARD4-AS1) on der5 (figure 1A, table 1). In addition, the CTNNA3 gene is disrupted by the insertion on der10 (figure 1A). CTNNA3 is associated with autosomal dominant cardiomyopathy, but our patient had no corresponding symptoms. Since none of the disrupted genes had been linked with polyposis, we focused on APC. Exact localisation of the breakpoints revealed that the open reading frame of APC was not disrupted by the rearrangement (figure 2). Instead, an upstream breakpoint at chr5: 112 714 041 (hg38) separated the promoter/exon 1B region from the remainder of the APC gene body moving it ~2 Mb towards the centromer and reversing its orientation (figure 2). The transcription of the APC gene from der10 would thus rely exclusively on promoter 1A. Both promoters are typically active with higher activity of promoter 1B compared with promoter 1A as shown, for example, by ~2 fold higher RNA polymerase 2 occupancy in the human colon cancer cell line HCT11631 (figure 2). In addition, both promoters are surrounded by an open chromatin structure and carry active chromatin marks (H3 acetylated on lysine 27, figure 2). Finally, both APC promoters reside in a common topologically associating domain (TAD) flanked by upstream and downstream CTCF boundary elements, suggesting that both are subject to the same regulatory inputs (figure 2). The rearrangement is predicted to disrupt colocalisation of both promoters in a common TAD thus potentially affecting APC mRNA expression.
Analysis of APC mRNA expressionTo determine possible consequences for APC mRNA expression, we performed long-read RNA sequencing on the Oxford Nanopore GridION platform. For this, we used a protocol merging Agilent’s SureSelectXT Low Input Target Enrichment System with Oxford Nanopore’s cDNA-PCR Barcoding Library Preparation Kit for highly efficient capture of 123 cancer-related transcripts.25 Total RNA was prepared from short-term cultures of the patient’s PBMCs. The translation inhibitor puromycin was added to one of two parallel cultures for 5 hours to inhibit nonsense-mediated mRNA decay (NMD).32
In triplicate experiments, we achieved a mean sequencing depth of APC mRNA of ~30 x. DGE analysis showed a reduction of APC mRNA by 38% in the patient’s PBMCs compared with 17 unaffected controls (figure 3A). This finding suggested an allelic reduction in APC expression. To confirm this, we assessed allelic mRNA expression based on synonymous single nucleotide variants (SNVs) located in APC (NM_000038.6) exons 12, 14 and 16. The patient’s genomic DNA isolated from PBMCs was heterozygous for these SNVs showing allelic ratios of ~50:50. At the mRNA level, the allelic ratios averaged over seven coding SNVs were shifted to ~25:75. This shift was maintained in PBMCs in which NMD was blocked, indicating that the allelic imbalance was due to changes in transcription rather than mRNA decay (figure 3B).
Assuming that the measured allelic imbalances reflect reduced expression of the rearranged APC allele on chromosome der10 and that the remaining wildtype allele retains its transcriptional activity at 100%, the reduction in total cellular APC mRNA calculated from RNA-seq based allele-specific quantification is ~33%. This number is in agreement with the 38% reduction in total APC mRNA measured by DGE analysis based on long-read RNA-seq (figure 3A). Finally, read density was selectively reduced in the promoter/exon 1B of the APC mRNA in the FAP patient relative to a control individual not afflicted by FAP (online supplemental figure S4A).
 Figure 4
Figure 4 Segregation of the genomic rearrangement in the patient’s offspring. PCR was performed on genomic DNA samples to amplify breakpoints 1, 6 and 9 shown in figure 1A. R=reference (unaffected individual), p=patient, #1, 2=progeny.
These results were independently confirmed by standard PCR amplification of APC exons 12 and 14 from cDNA derived from the patient. For both SNVs, exon 12 c.1458T>C p.(Tyr486=) and exon 14 c.1635G>A p.(Ala545=), the genomic allele balance of ~50:50 was shifted to ~25:75 in cDNA prepared from cultured PBMCs (figure 3C). Allelic imbalances were even more pronounced when cDNA was prepared directly from whole blood, suggesting that stimulation of cell proliferation by short-term culture may restore some residual activity of the rearranged APC allele and thus may lead to overestimation of APC mRNA levels in the patient’s tissues (figure 3C).
Consistent with separation of the APC promoter/exon 1B from the remainder of the APC gene and its insertion in reverse orientation upstream of EPB41L4A (figure 1A), RNA-seq analysis revealed a fusion transcript linking APC exon 1B with EPB41L4A (NM_022140.5) exon 2 (online supplemental figure S4B). This fusion transcript further confirms the structural rearrangement and indicates that the mislocalised APC promoter 1B retains activity in PBMCs.
Segregation analysisThe patient’s father who died from CRC at an age between 70 and 75 was tested negative for the genomic rearrangement by targeted PCR analyses (data not shown). No material was available of the patient’s mother who died of CRC in her mid-40s. Two of the patient’s healthy children were assessed and tested negative by PCR for three breakpoints of the rearrangement and could thus be spared extensive surveillance measures (figure 4). The majority of de novo chromosomal structural aberrations found at term is known to be of paternal origin (reviewed in ref 33). To assign the parental origin of the patient’s rearranged APC allele, we performed Sanger sequencing of the paternal APC gene. The father was found homozygous for the variant c.1635G>A (data not shown), which is diagnostic of the patient’s intact APC allele based on its higher allelic mRNA expression (online supplemental table S2). Thus, the allele that underwent rearrangement in the patient was derived from the mother.
DiscussionComprehensive genomic analysis employing chromosomal microarray profiling, optical mapping (Bionano Genomics), long-read GS and RNA-Seq combined with FISH and standard PCR was able to unravel a FAP patient, which had gone unsolved for nearly 20 years. The complex rearrangement we identified involves 14 fragments from chromosome 5 of which 10 reinserted into chromosome 10 in a seemingly random order and orientation, whereas three were lost and one re-inserted into der5. As such, the rearrangement fulfils the major criteria of constitutional chromothripsis19 with marked clustering of breakpoints within a 3.9 Mb segment and interspersed loss and retention of heterozygosity affecting only one haplotype. Analysis of the break point junctions revealed small insertions and deletions but limited homology (online supplemental table S3), indicating random fusion of shattered chromosomal fragments by non-homologous end joining.34 Our analysis suggests a local genotoxic insult imparted on chromosome 5q22.1q22.3 accompanied by a singular DNA strand break in chromosome 10 into which the chromosome 5 fragments randomly inserted. Recent in situ genome sequencing demonstrated close spatial proximity of chromosomes 5 and 10 in nuclei of human diploid PNP1f fibroblasts,35 raising the intriguing possibility that integration of chromosome 5 fragments into chromosome 10 was due to physical juxtaposition of the respective chromosomal territories.
The genotoxic insult leading to chromothripsis may have occurred in the parents’ germ cells or during the patient’s early embryonic development. Constitutional chromothripsis is known to occur preferentially in the male germline,33 but we have found that the APC allele involved in the rearrangement was of maternal origin. Considering the rareness of female germline chromothripsis, the event described here most likely occurred during fertilisation or shortly thereafter in the early zygote. Alternatively, the patient may have inherited the rearrangements from the mother. No material was available from the deceased mother who was diagnosed with CRC in her mid-40s, while a diagnosis of FAP was not confirmed. Parental transmission of a rearrangement of the described complexity might result in unbalanced transmissions but neither the patient nor the mother reported children with congenital malformations and developmental delay or miscarriages in the family. In addition, the rearrangement was not passed on to both of the patient’s children that were available for testing, possibly suggesting that unbalanced transmission might interfere with normal germ cell maturation or very early embryonic development (figure 4). Taken together, the rearrangement most likely occurred de novo in the patient during early embryogenesis.
Our data show that spatial separation of promoter/exon 1B from the coding region of APC coincides with a 30%–40% reduction in APC mRNA expression (figure 3). Considering that complete loss of one allele would reduce mRNA expression by 50%, promoter 1A, which remained intact on the rearranged allele, appears to make only minor contributions to APC mRNA expression. This is consistent with the previous demonstration that APC promoter 1B is dominant over 1A and that germline deletions of promoter 1B are pathogenic.8 36–38 Thus, reduced expression of APC mRNA may underlie the patient’s polyposis.
Whereas APC promoter 1B germline deletions have only been found in classical FAP,7 the patient studied here has attenuated FAP. It is thus possible that some of the other genes disrupted through the rearrangement modify the phenotype caused by the promoter 1B alteration. Notably, the rearrangement is predicted to disrupt three other genes that have been implicated in the APC–Axin–GSK3B–β-catenin signalling pathway. The gene products of MCC (mutated in colon cancer), a colon tumour suppressor, as well as CTNNA3 were shown to suppress β-catenin-dependent transcription in SW480 colon cancer cells,39 40 while EPB41L4A is a transcriptional downstream target of β-catenin/TCF41 that has been linked with colon cancer.42 Although it is difficult to exactly predict the sum outcome of the alterations to the β-catenin pathway conferred by the complex rearrangement, the combined effects may modify the penetrance of the patient’s specific pathogenic APC variant.
In conclusion, our study showcases the power of multilevel genomic analysis in resolving pathogenic variants underlying FAP that escaped routine diagnostic approaches including NGS. Within our workflow, optical mapping appears a sensible first approach to delineate large-scale structural variants at kilobase pair resolution. This can be complemented by low-depth long-read GS for fine mapping of breakpoints at nucleotide resolution thus providing information for the development of low-cost PCR assays for cascade testing. This workflow proved efficient in unravelling constitutional chromothripsis affecting chromosome 5q22.1q22.3 as a previously unidentified pathogenic mechanism in FAP that may also underlie other undiagnosed cases of FAP as well as other hereditary monogenetic diseases.
Data availability statementData are available on reasonable request.
Ethics statementsPatient consent for publicationEthics approvalPatients and healthy control subjects were recruited from our clinical practice (MGZ Medical Genetics Center Munich) according to rules and guidelines approved by the Institutional Review Board of the Ludwig-Maximilians-University (Protocol Number 17-805). Written, informed consent was obtained from all participants in this study before sample and data collection. The study adhered to the principles set out in the Declaration of Helsinki.
AcknowledgmentsWe thank all MGZ lab and medical staff for supporting this work.
留言 (0)